Liposome Drug Products Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability; and Labeling Documentation Guidance…

ADC Linkers: An Assurance for Controlled Delivery of Antibody Drug Conjugate
This article belongs to the Special Issue Chemically Enhanced Peptide and Protein Therapeutics. Information about the original article, authors, and research team can be found below. If you’re in need of high-purity PEGs and ADC linkers for your research, browse our collection of over 7,000 PEG products online or contact our team for custom PEG synthesis, PEGylation, bulk orders or other inquiries.
Table of Contents
Abstract
As one of the major therapeutic options for cancer treatment, chemotherapy has limited selectivity against cancer cells. Consequently, this therapeutic strategy offers a small therapeutic window with potentially high toxicity and thus limited efficacy of doses that can be tolerated by patients. Antibody-drug conjugates (ADCs) are an emerging class of anti-cancer therapeutic drugs that can deliver highly cytotoxic molecules directly to cancer cells. To date, twelve ADCs have received market approval, with several others in clinical stages. ADCs have become a powerful class of therapeutic agents in oncology and hematology. ADCs consist of recombinant monoclonal antibodies that are covalently bound to cytotoxic chemicals via synthetic linkers. The linker has a key role in ADC outcomes because its characteristics substantially impact the therapeutic index efficacy and pharmacokinetics of these drugs. Stable linkers and ADCs can maintain antibody concentration in blood circulation, and they do not release the cytotoxic drug before it reaches its target, thus resulting in minimum off-target effects. The linkers used in ADC development can be classified as cleavable and non-cleavable. The former, in turn, can be grouped into three types: hydrazone, disulfide, or peptide linkers. In this review, we highlight the various linkers used in ADC development and their design strategy, release mechanisms, and future perspectives.
1. Introduction
Cancer is a serious life-threatening disease [1] causing over 8 million deaths worldwide each year [2]. This disease is commonly treated by surgery [3], radiotherapy [4] and chemotherapy [5,6], the latter being the treatment most widely used due to its capacity to target rapidly dividing cancer cells [2]. However, the use of chemotherapeutic drugs faces constant limitations in terms of non-specificity, meaning that they kill not only the tumor cells but also healthy cells and cause serious adverse reactions, narrow therapeutic windows, and increased drug resistance [7,8,9,10,11,12,13]. Targeted drug therapy could potentially address these challenges, as it facilitates the delivery of drug agents to unhealthy cells without harming healthy ones [14,15,16,17,18,19,20]. Antibodies are a rapidly growing class of drug that play a major role in human health, mostly in oncology, autoimmunity and chronic inflammatory diseases [21,22,23].
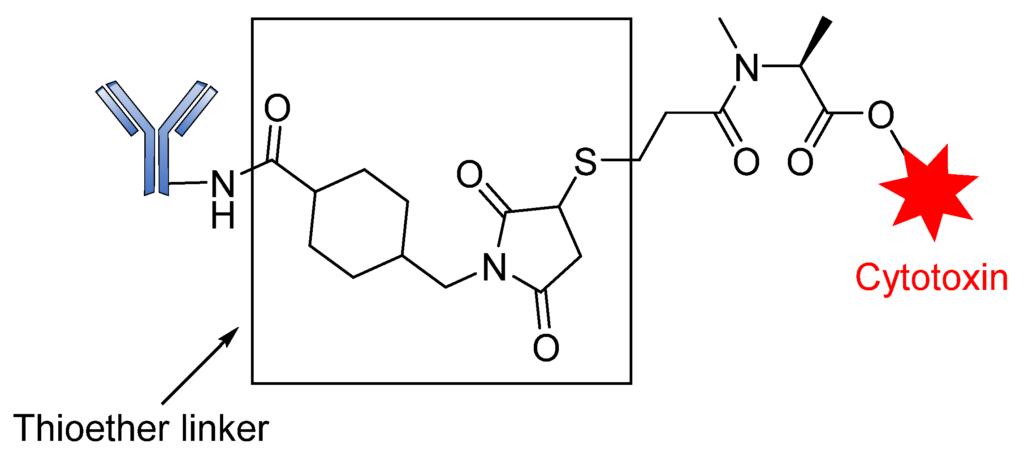
The development of antibody drug conjugates (ADCs) as targeted drug therapies has made significant progress over the last century [24,25,26,27]. In 1897, Paul Ehrlich, a German scientist, was the first to propose the ‘magic bullet’ theory for delivering toxic compounds to unhealthy cells. His idea later evolved to become what is today known as ADCs [28,29,30,31]. This class of therapeutic agents consists of recombinant monoclonal antibodies (mAbs) (which direct the drug to the target cells) that are covalently bound to cytotoxic chemicals (known as warheads) via synthetic linkers, as shown in Figure 1. ADCs offer the prospect of delivering a toxic payload directly to a target, with minimal off-target toxicity [32].
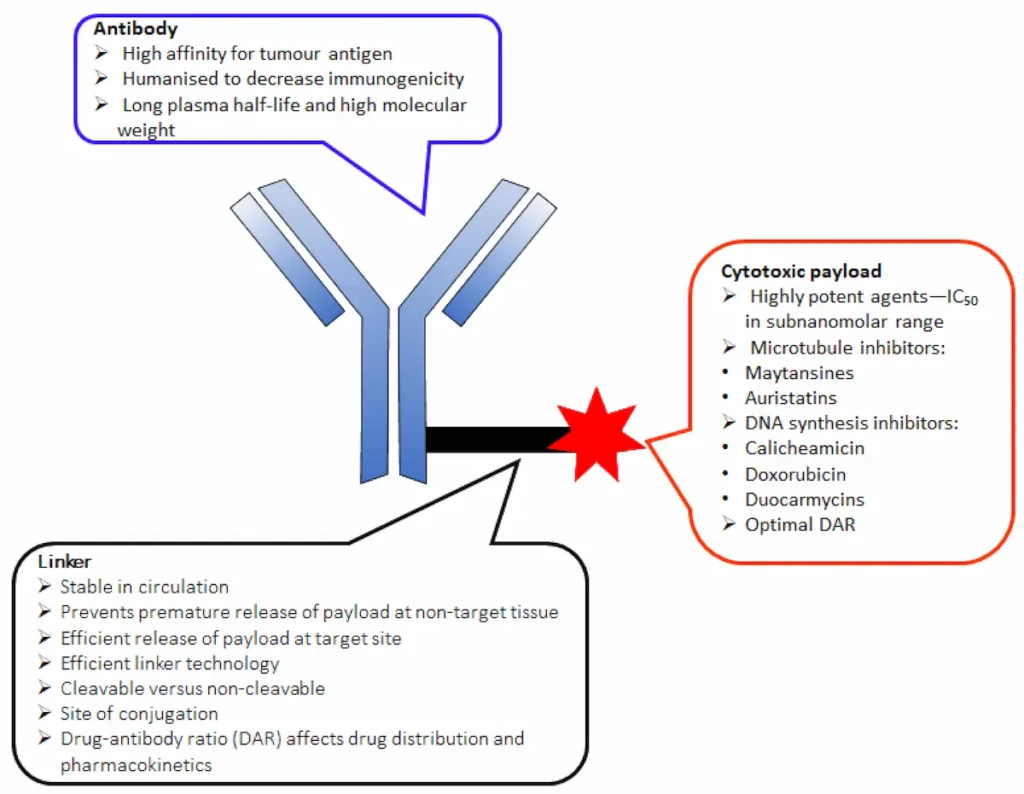
Two main factors determine the successful development of an ADC. First, the choice of linker is crucial, as it accounts for: (i) high plasma stability in circulation to prevent premature drug release (t1/2 > 1 week); (ii) maintenance of the properties of the mAb and cell-killing ability of the cytotoxic drugs, along with a reduction of systemic toxicity; (iii) high aqueous solubility that allows bioconjugation of lipophilic drugs and prevents antibody aggregation; and (iv) drug release in the right circumstances to maximize the therapeutic effect [33,34,35,36,37,38]. Second, successful ADC development is also dependent on the drug-antibody ratio (DAR), which is the number of drug molecules attached to the antibody via a linker. A low DAR decreases ADC efficacy, while a high DAR often results in ADC instability, increased systemic effect, and reduced half-life, and it alters the pharmacokinetic properties of the molecule [26]. However, the choice of appropriate linker depends on the functional groups present in the mAbs and cytotoxic drugs. This review provides an update of FDA-approved ADCs and the linkers used in the design of these drugs.
2. The Key Components of an ADC
2.1. Monoclonal Antibody (mAb)
An important component of an ADC is the antibody. The basic premise for the selection of an antibody for ADC design is its ability to specifically identify and bind to a well-characterized tumor antigen–receptor and deliver the payload to the tumor cell in the process. The antibody must also have high binding affinity to the specific target antigen, low immunogenicity, appreciable stability in the bloodstream, and low cross-reactivity [2,35]. Most antibodies used in ADC design are selected from human immunoglobulin G (IgG) subclasses (IgG1, IgG2, IgG4), which consist of tw heavy and two light chains [20,26,33]. ADC-targeted antigens must be highly expressed on the tumor cells [39] and should also have internalization properties to enhance the receptor-mediated endocytosis of the ADC [40]. Currently, Nectin4, CD79b, CD22, CD33, HER2, CD30, FOLR1, and TROP2 are the most targeted antigens in ADCs. In addition, over 70 other antigens are in different stages of clinical development [41,42,43,44].
2.2. Cytotoxic Drug
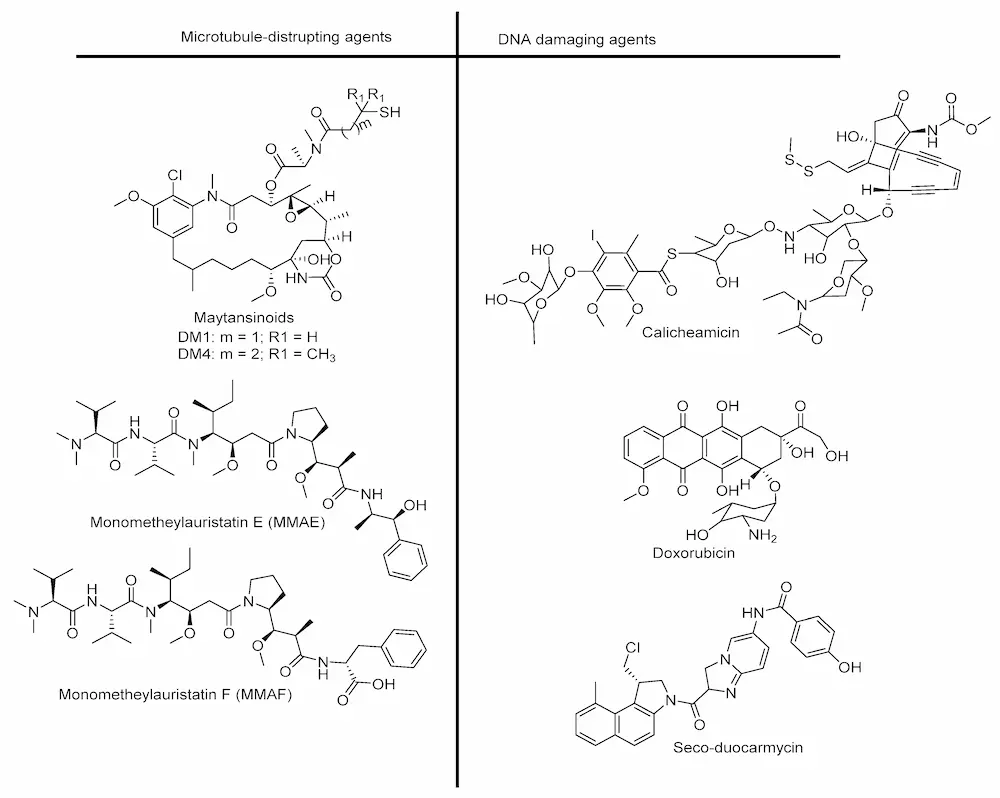
Cytotoxic drugs are highly potent agents used to kill cancer cells. They can prevent cell division either by disrupting microtubule assembly, thus inhibiting mitosis, or by binding to the minor groove of DNA, leading to the cleavage of double-strand DNA. The latter process causes cell death/apoptosis [45,46]. Therefore, a cytotoxin is required to have maximum plasma stability and an in vitro subnanomolar IC50 (half maximal inhibitory concentration) value for tumor cells, as only 1–2% of injected ADCs reach the tumor [33,47]. Cytotoxic drugs are transported in the bloodstream throughout the body. At present, auristatins and maytansinoids are the drugs most frequently used for ADC development (see Figure 2 for their chemical structures) [37]. The conjugation of these drugs to mAbs is usually achieved via a chemical linker attached to the thiol residue of Cys or to the amino group of Lys antibody molecule. Several drugs used in the design of ADCs are listed in Figure 2.
3. Linker Chemistry and Conjugation to Antibody
The linker, which is the focus of this review, is an essential component in ADC design. It connects the antibody to the cytotoxic payload via covalent conjugation [26,48,49,50]. The key requirement of a linker is that it must ensure chemical stability of the ADC within the bloodstream (i.e., have a half-life 10 times longer than the ADC) and allow for rapid release of the payload at the target site after internalization [51,52]. In addition to the above parameters that minimize premature drug release [39], hydro/lipophilicity, a property that enhances the coupling of payloads and reduces immunogenicity, is also a key aspect of linkers [53,54].
Two types of linkers, namely cleavable and non-cleavable, are used in ADC development (Figure 3). These linkers play major roles in determining pharmacokinetic properties, selectivity, therapeutic index, and the overall success of the ADC. With the development of ADCs, a series of linkers have been exploited [37]. Cleavable and non-cleavable linkers have been proven to be safe in preclinical and clinical trials. Linkers are broadly classified on the basis of the drug release mechanism and their stability in circulation [55,56].
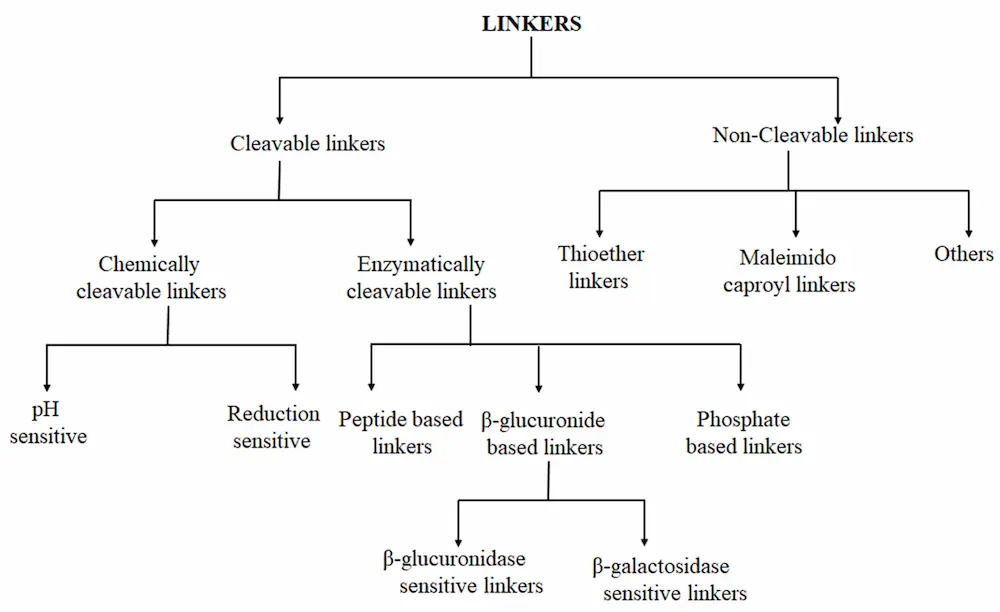
3.1. Cleavable Linkers
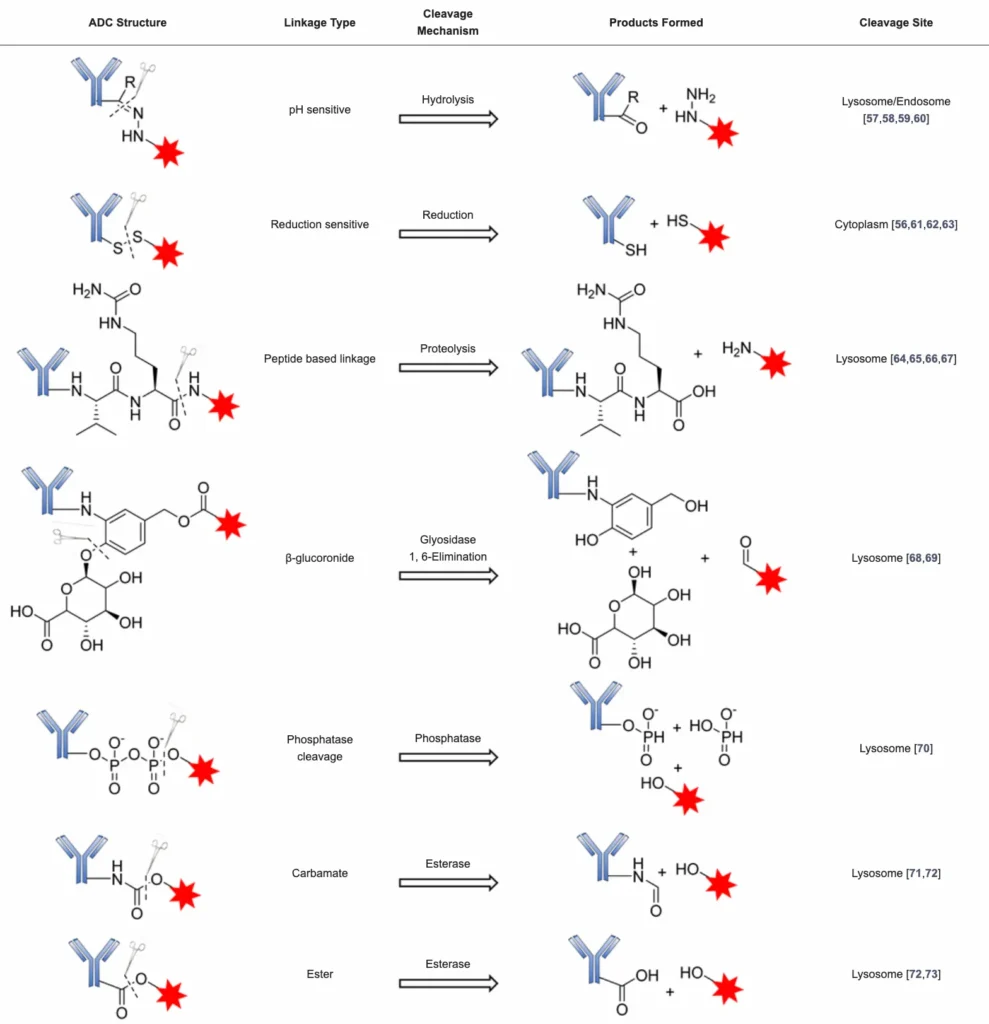
Cleavable linkers play a pivotal role in the success of ADCs. They are stable in blood circulation for a long time and efficiently release their payload in the tumor microenvironment. Some of the cleavable linker strategies available and cleavage conditions are summarized in Table 1 and are subsequently discussed.
3.1.1. Chemically Labile Linkers
ADCs containing chemically labile linkers, including acid-cleavable linkers and reducible linkers, take advantage of the differential properties between plasma and the cytoplasmic compartments to release the payload.
Acid-Cleavable/pH-Sensitive Linkers
This class of linkers is stable to alkaline environments but highly sensitive to acidic environments such as the hydrazone. They take advantage of the low pH of the endosome (pH = 5–6) and the lysosome (pH = 4.8) to trigger the hydrolysis of the acid-labile hydrazone linker and subsequently release the payload [48].
BR96-Doxorobicin (BR96-Dox) is a good example of a drug conjugate constructed using an acid-sensitive linker. This ADC was designed by linking doxorubicin, an intercalating agent that blocks DNA replication over a (6-maleimidocaproyl) hydrazone linker bonded to cysteine residues of humanized BR96 monoclonal antibody (Figure 4) [74]. The preclinical results of BR96-Dox were remarkable due to its potential to deliver high doses of doxorubicin to tumors. These doses were found to be capable of curing hypodermic human breast, and lung and colon tumors [75,76]. Although BR96-Dox has a high DAR, it showed insufficient potency in clinical trials. In addition, the half-life of the drug was too short compared to that of the naked BR96 mAb in humans [77].
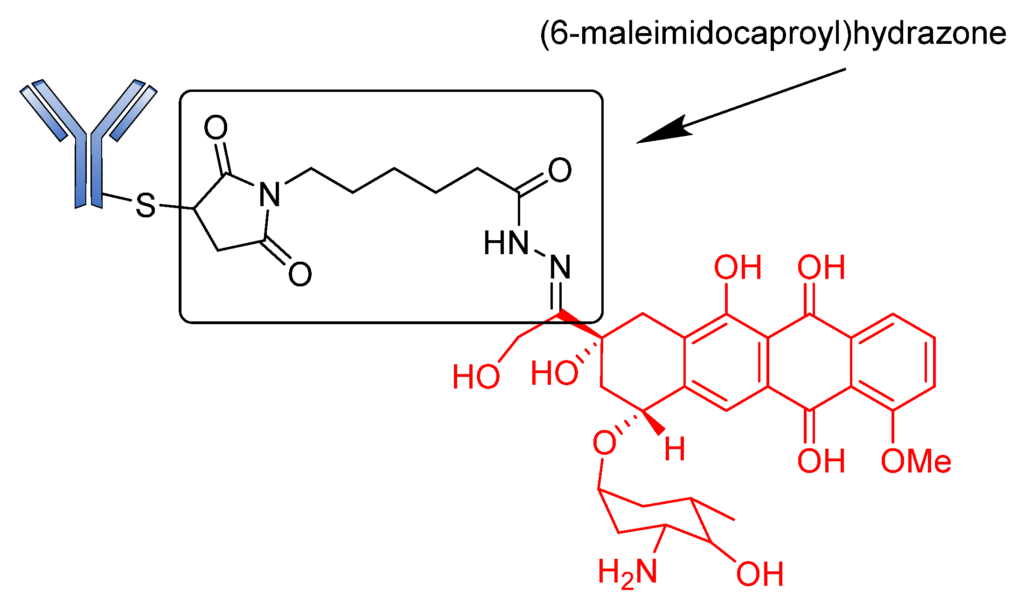
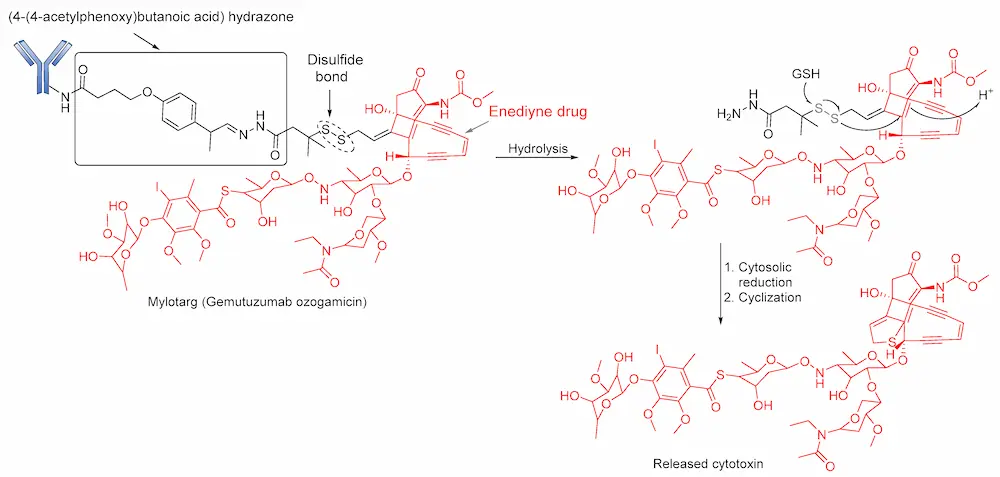
Hydrazone linkers were also explored in the design of Gemtuzumab ozogamicin (Mylotarg) and Inotuzumab ozogamicin (InO; Besponsa). Mylotarg, the first ADC to gain regulatory approval, is used for the treatment of patients suffering from relapsed acute myelocytic leukemia (AML). This ADC consists of N-acetyl-γ-calicheamicin (a DNA- damaging agent) covalently attached to humanized anti-CD33 antibody (hP67.6) through an acid-cleavable hydrazone linker [78]. The 4-(4-acetylphenoxy) butanoic acid moiety permits conjugation to Lys residues of the hP67.6 antibody over an amide bond and forms an acyl hydrazone linkage with N-acetyl-γ-calicheamicin dimethyl hydrazide. Upon internalization of the ADC, the calicheamicin prodrug is released by hydrolysis of the hydrazone into the lysosomal compartment of CD33-positive tumor cells. The DNA-damaging agent (enediyne drug) is then activated by reduction of the disulfide bond (Figure 5). However, the stability of this bond has been enhanced by the introduction of two methyl groups to the α-carbon bearing the disulfide bonds to prevent premature release of the calicheamicin metabolite [77]. Initial clinical studies of Mylotarg in relapsed patients led to its approval in 2000. However, the instability of the linker and heterogeneous nature of the conjugate caused the premature release of the drug before it reached its target site [79] and, consequently, the ADC was voluntarily withdrawn from the market by the US FDA in 2010 [80]. However, this ADC was later reapproved for use in 2017 when its benefits were considered to outweigh its risks [19].
Similarly, collaborative work between Wyeth and Celltech led to the development of Inotuzumab ozogamicin (CMC-544, Besponsa), a calicheamicin-based ADC that consists of a recombinant humanized anti-CD22 antibody attached to N-acetyl-γ-calicheamicin dimethyl hydrazide via the acid-labile (4-(4′-acetylphenoxy)butanoic acid) hydrazone linker. Although this ADC is closely related to Mylotarg, it showed more stability in both human plasma and serum (rate of hydrolysis of 1.5–2%/day over 4 days) [81,82]. Used to treat relapsed B-cell precursor acute lymphoblastic leukemia, Besponsa differs from Mylotarg in that it targets CD22-bearing antigen cells.
Reducible Linkers
Reducible or glutathione-sensitive disulfide linkers are alternatives to acid-labile hydrazone linkers in ADC design. Disulfide bonds are comparatively stable in circulation yet are reductively cleaved by intracellular glutathione to release the payload [26]. Glutathione is a low molecular weight thiol present in the cytoplasm (1–10 mmol/L) and extracellular environment (2–20 µmol/L in plasma) [6,20,83,84]. The basic premise for the cleavage of ADCs containing these linkers is the difference in reduction potential in the intracellular compartment as opposed to plasma, bearing in mind that glutathione is highly released during cell replication; hence, a high concentration of glutathione can be found in cancer cells [85]. These linkers generate a neutral payload that can diffuse into neighboring cancer cells and produce bystander-killing effects [86].
As earlier discussed, disulfide linkers have found significant clinical applications from their combination with hydrazone in the development of Pfizer’s Mylotarg and Besponsa. Remarkably, Immunogen reported the fortuitous development of some disulfide-based ADCs: SAR3419 (antiCD19 maytansine conjugate), IMGN901 (anti-CD56 maytansine conjugate), and AVE9633 (anti-CD33 maytansine conjugate).
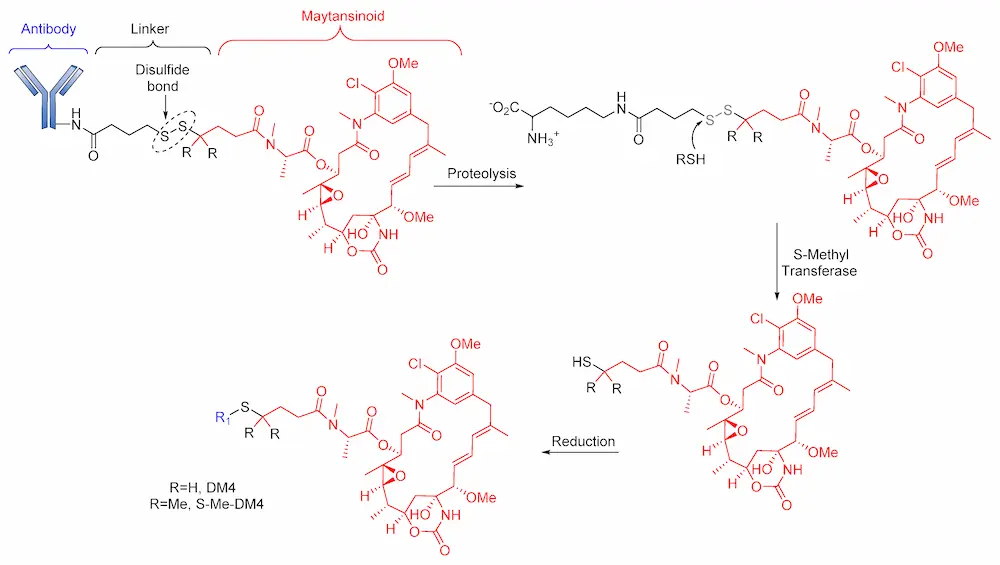
Notably, in 2011, Kellogg et al. reported the synthesis of huC242-SPDB-DM4 (IMGN242), a disulfide-containing ADC [87], comprises huC242 antibody linked to tubulin. This ADC inhibits cytotoxin (maytansinoids, DM4) via a disulfide linker with varying levels of steric hindrance around the disulfide (Figure 6) [87]. Unlike its non-cleavable counterpart (huC242-SMCC-DM1), huC242-SPDB-DM4 has a DAR > 4, conferring it with higher in vitro activity. The study of this ADC also revealed its high stability to dithiothreitol reduction in isolated plasma of CD1 mice. This drug conjugate, with intermediate disulfide bond stability and two methyl groups on the maytansinoid side of the bond and no methyl groups on the linker side of the bond, showed a better efficacy than huC242-SMCC-DM1 [87]. An in vivo study of this maytansinoid conjugate also revealed its effectiveness in killing neighboring non-targeted cancer cells as a result of on-target cleavage of the cytotoxic metabolites, which diffuse into neighboring cells to elicit a bystander effect.
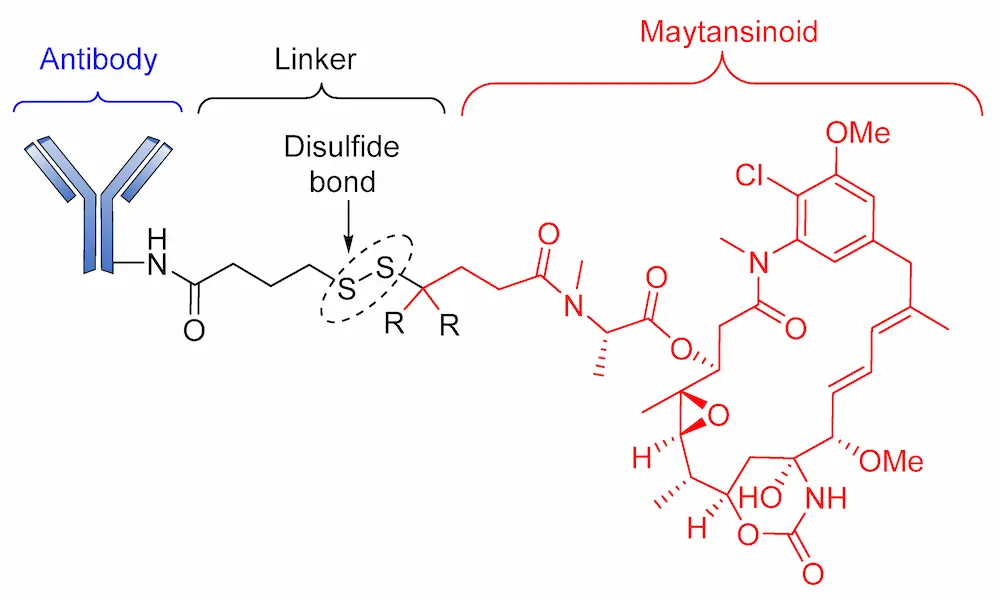
Although Lorvotuzumab mertansine (IMGN901) is still in phase II clinical trials, researchers at Immunogen have reported an anti-CD56 effect. IMGN901 consists of a potent maytansinoid attached to a CD56-binding monoclonal antibody through a disulfide linker (Figure 7) [88].
3.1.2. Enzyme-Cleavable Linkers
Chemically labile linkers often have limited plasma stability, thereby leading to premature drug release. In this regard, as an alternative strategy, enzyme-cleavable linkers have achieved clinical success in controlled drug release. Unlike chemically labile linkers, enzyme-cleavable ones take advantage of the high concentration of unique hydrolytic enzymes in cellular compartments to degrade peptides and carbohydrates.
Peptide-Based Linkers
Peptide-based linkers, also known as lysosomal protease-sensitive linkers, such as valine–citrulline (Val–Cit), phenylalanine–lysine (Phe–Lys), and valine–alanine (Val–Ala) dipeptide linkers, are the most widely used linkers in ADC design. This strategy utilizes intracellular protease, such as Cathepsin B, which recognizes and cleaves a dipeptide bond, thus leading to the release of the cytotoxic drugs [89]. Due to unsuitable pH conditions and serum protease inhibitors, peptide-based linkers show greater systemic stability, with rapid enzymatic release of the payload in the target cell [90]. Exploring these types of linkers in ADC development often requires a conjugating spacer molecule due to the bulky nature of payload. The reagent most commonly used for this purpose is para-aminobenzyl carbamate (PABC) (Figure 8), which shows self-cleavage capacity, thereby facilitating the release of the unmodified payload [70].
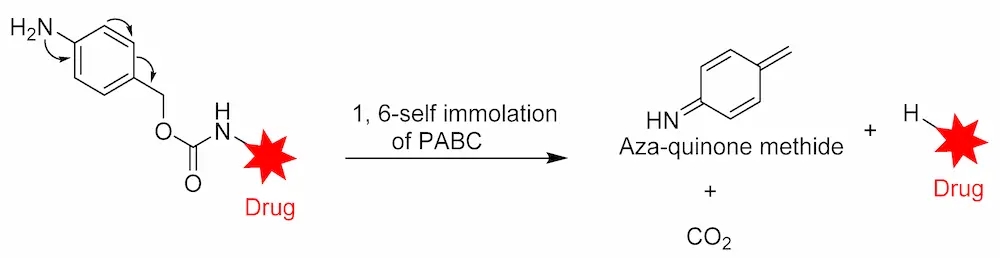
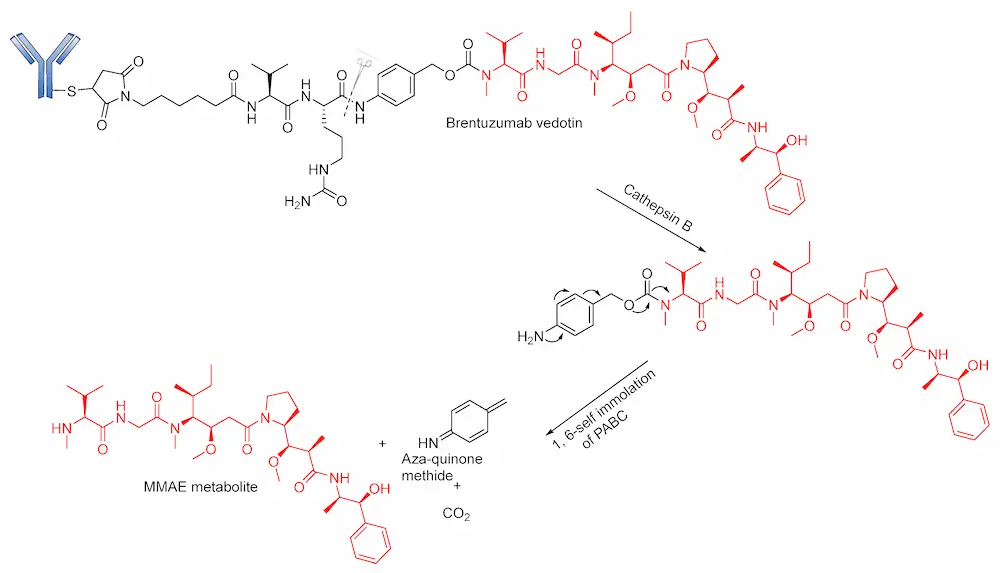
Developed by Seattle Genetics/Millennium, FDA-approved Brentuximab vedotin (BV; Adcetris) is a good example of an ADC in which this linker has been explored with marked clinical success. BV consists of MMAE conjugated to an anti-CD30 antibody via a self- immolative protease (cathepsin B-sensitive) Val–Cit- para-aminobenzyl carbamate (PABC) linker [20,55]. BV is internalized into CD30-expressing cells, and the dipeptide bond undergoes proteolytic cleavage followed by self-immolation via a 1,6 elimination of PABC to release MMAE (Figure 9). Although ADCs containing this linker are generally stable in physiological conditions, an unidentified serine protease is known to cleave the linker in mouse plasma [20,91]. BV gained accelerated approval in 2011; however, due to several adverse effects (neuropathy, neutropenia, anemia and thrombocytopenia), its approval was halted [92,93]. However, after a series of modifications, it was fully approved in 2015, and it is considered suitable for the treatment of Hodgkin’s lymphoma [94,95].
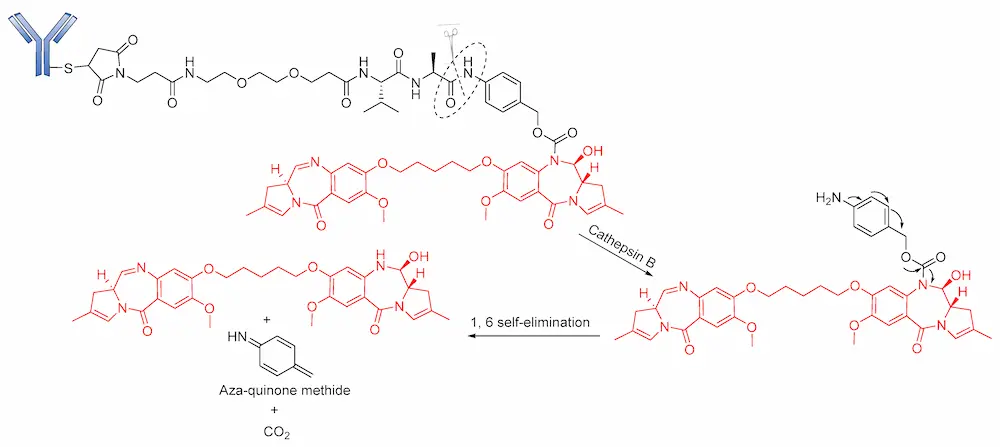
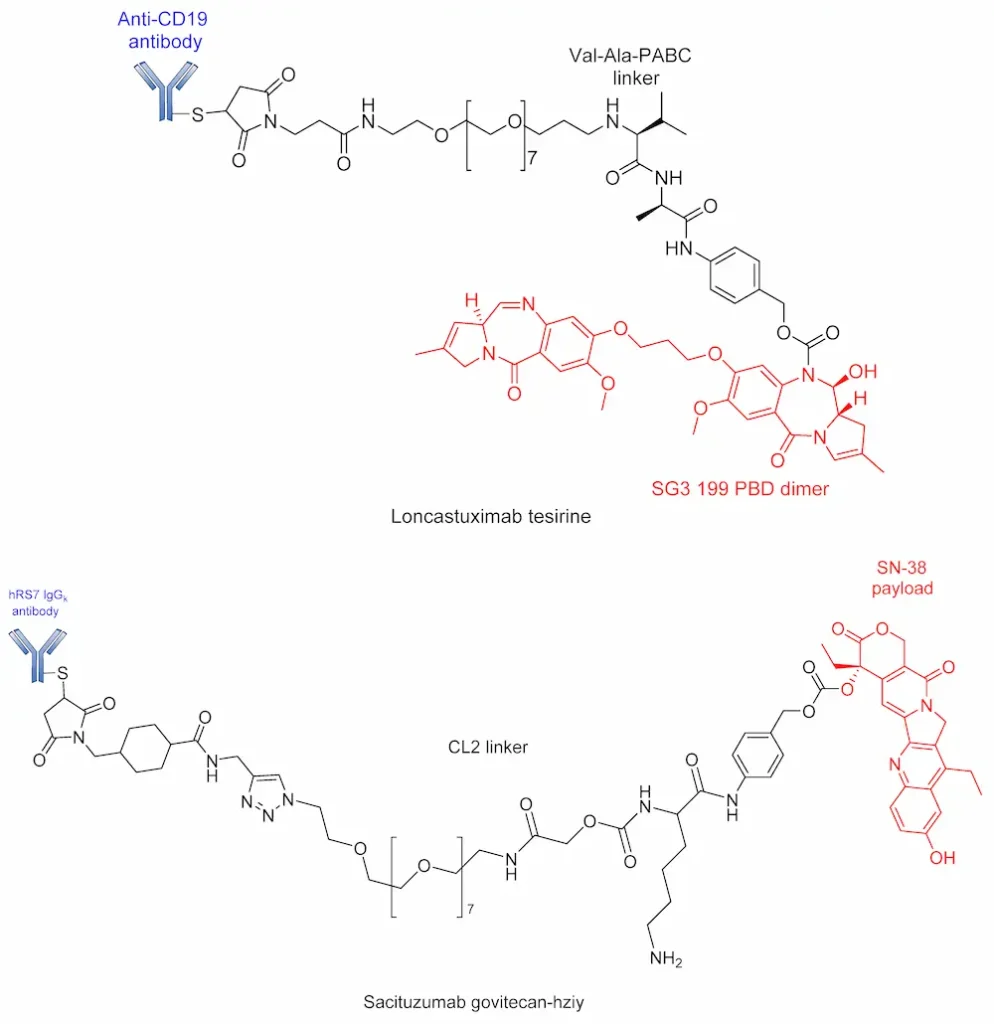
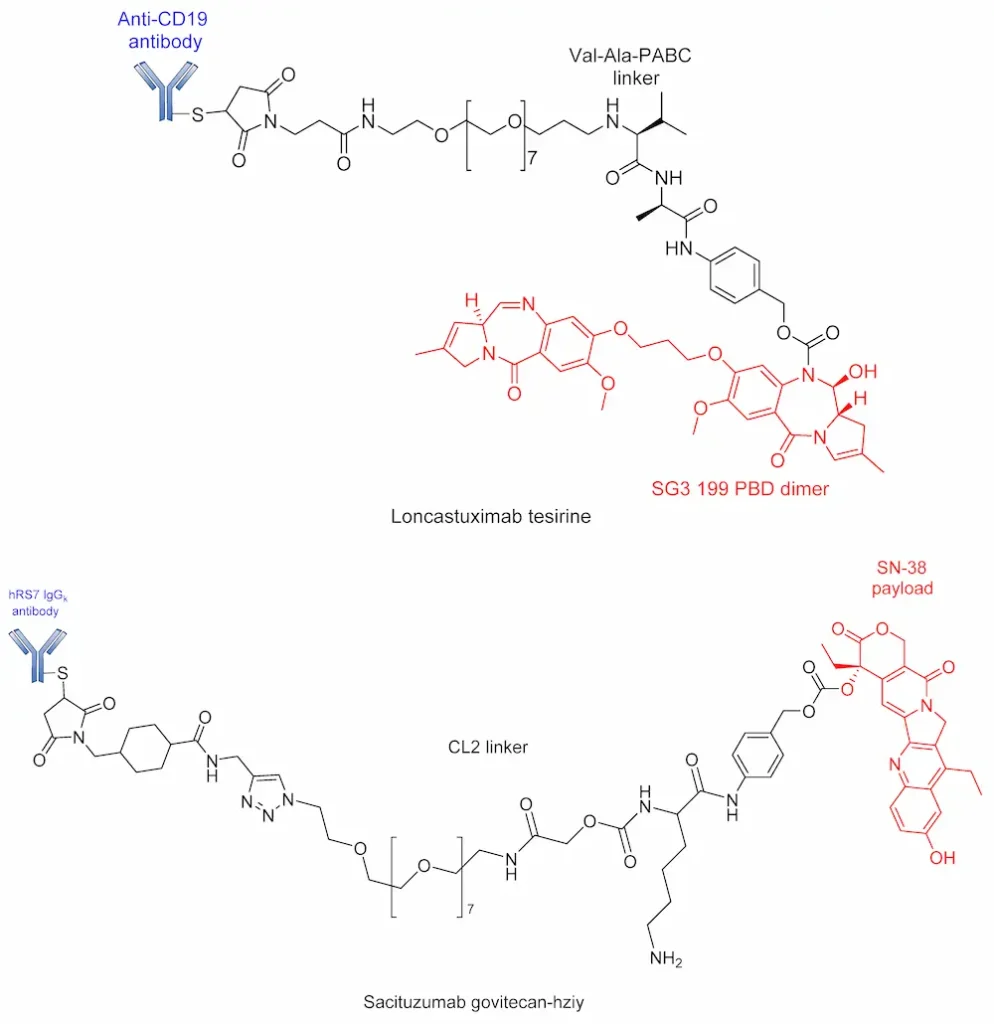
An alternative to the Val–Cit linker is the protease-cleavable Val–Ala dipeptide linker, which is being used in the development of many pyrrolobenzodiazepine-containing ADCs. Currently in phase III clinical trials, Rovalpituzumab tesirine (Rova-T; SC16LD6.5) is a biomarker-specific ADC for the exclusive targeting small-cell lung cancers expressing Delta-like protein 3 (DLL3) antigen [96]. Rova-T is made up of SC16 antibody conjugated to a pyrrolobenzodiazepine (PBD) payload via a PEG8 spacer, namely a Val–Ala linker. However, the bulky nature of the drug calls for the use of a self-immolative PABC to afford the straightforward release of the payload. Similar to BV, Rova-T first undergoes proteolytic cleavage of the Val–Ala linker followed by self-immolation of PABC to release the PBD payload (Figure 10) [97].
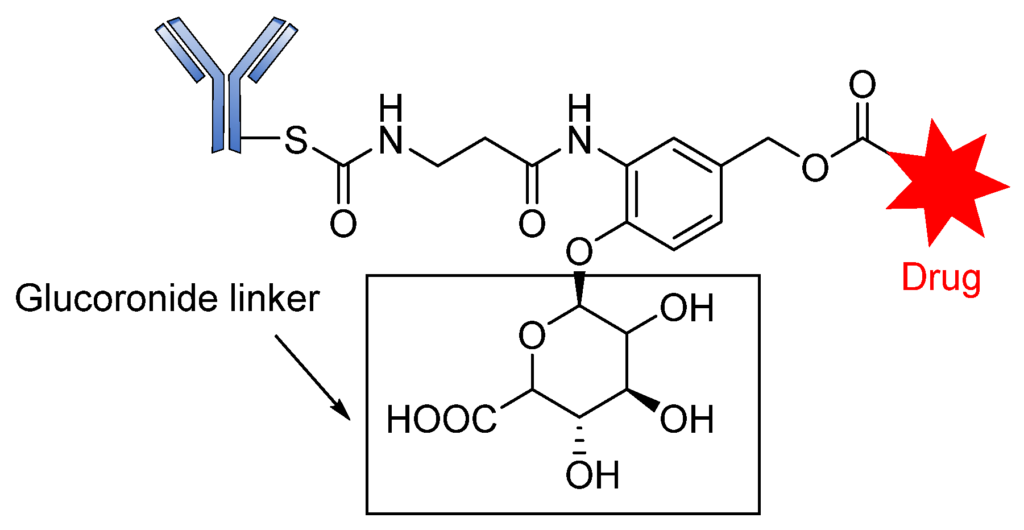
Glycosidase-Sensitive Linkers
β-Glucuronidase-cleavable linkers
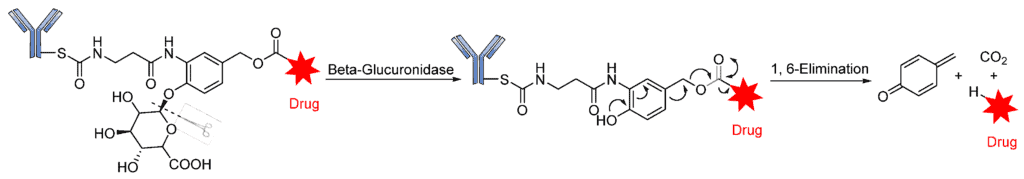
Glycosidases, such as β-glucuronidases, are a class of hydrolytic lysosomal enzymes that degrade β-glucuronic acid residues into polysaccharides. They are found exclusively in the lysosomal compartment of the cell, and they work under hydrophilic environments to release payloads from conjugates. Similar to cathepsin B, β-glucuronidases are secreted in the necrotic areas of some tumors. Remarkably, these molecules are enzymatically active in the extracellular environment. Given this property, in 1988, Tietze et al. addressed for the first time the activity of β-glucuronidase-responsive prodrugs on neighboring tumor cells [69].
Moreover, in a comprehensive study, Jeffrey and coworkers evaluated the properties of β-glucuronic-based acid linkers ADCs. Using highly potent microtube inhibitors (MMAE and MMAF) and a DNA-damaging agent (doxorubicin), β-glucuronidase-susceptible ADCs were designed by covalent conjugation of the cytotoxic agents to the antibody via a β-glucuronide linker attached to a self-immolative PABC spacer molecule (Figure 11) [98].
The introduction of the self-immolative spacer enhances linker stability and allows safe release of the potent cytotoxin [99]. The resultant β-glucuronide MMAF drug conjugates gave a DAR of 8.3 with low levels of aggregation and a half-life of 81 days, far greater than that of the dipeptide linkers. Apart from the use of auristatins and doxorubicin, this strategy has also been employed in targeting special classes of cytotoxic agents such as anthracyclines, camptothecin derivatives, taxanes, hedgehog inhibitors, nitrogen mustards, and histone deacetylase inhibitors [69]. Most of these drug conjugates contain the self- immolative PABC spacer molecule between the β-glucuronide linker and the drug moiety, thereby offering straightforward release of the payloads upon internalization [69]. The release of the drug therefore proceeds in two steps: (i) the enzymatic hydrolysis of the glycosidic bound; and (ii) the spontaneous decomposition of the linker, leading to the release of the active compound (Figure 12) [70].
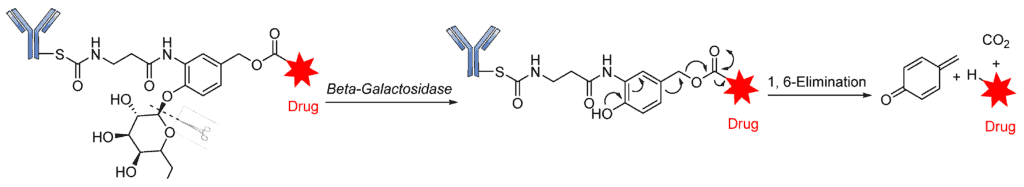
β-Galactosidase-cleavable linkers
Another class of hydrolytic lysosomal enzymes is β-galactosidase, which degrades β-glycosidic linkage formed between a galactose and its organic moiety. Kolodych and coworkers recently described both the in vitro and in vivo activities of β-galactosidase-cleavable ADCs. The study revealed that galactosidase-based drug conjugates have greater therapeutic efficacy in isolated mouse plasma compared to the approved Trastuzumab emtansine used for the treatment of breast cancer [100]. Similar to β-glucuronidase, β-galactosidase is overexpressed in certain tumors, where it hydrolyzes β-galactoside (Figure 13).
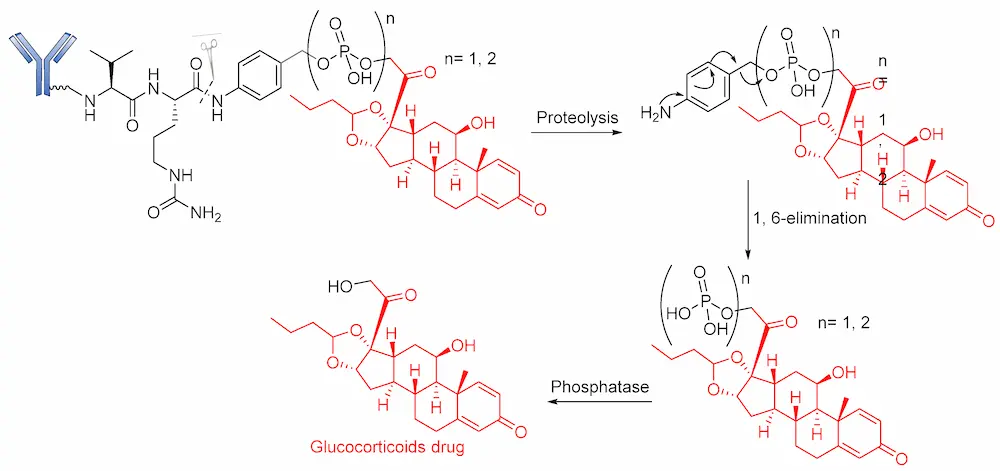
Phosphatase-Cleavable Linkers
These belong to another vital class of enzyme-cleavable linkers expressed exclusively to target enzymes in the lysosomal compartment. These linkers target pyrophosphatase and acid phosphatase enzymes, which hydrolyze pyrophosphates and terminal monophosphates into their respective alcohols. Kern and coworkers reported the usefulness of a phosphate-bridged Cathepsin B-sensitive linker in delivering glucocorticoids to tumor cells [101]. By using the PABC spacer molecule, they developed an aqueous, soluble phosphate drug conjugate comprising the Val–Cit-PABC cleavable linker. Upon proteolytic cleavage of the dipeptide linker (Val–Cit), PABC is self-eliminated, leading to the hydrolysis of the terminal phosphate by phosphatase to release the payload (Figure 14). An in vitro study of ADCs containing this linker revealed that they showed high blood stability, rapid lysosomal cleavage, and aqueous solubility. While these properties thus support the applicability of this linker in bioconjugation and ADCs containing lipophilic payloads, there is no in vivo proof to validate this approach. Moreover, this linker system increases the space of ADC payload options by exploiting the attachment of a payload via the aliphatic alcohol of the phosphate group.
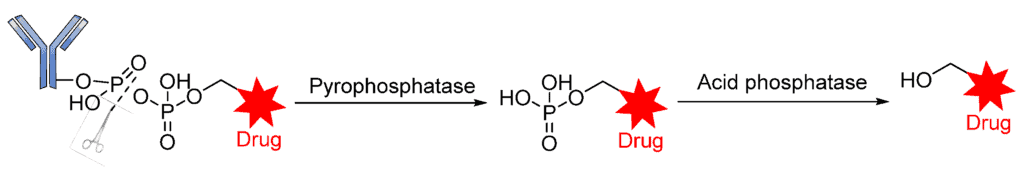
Recently, in similar but more elaborate research, Kern and coworkers reported the synthesis of highly soluble pyrophosphate-containing ADCs for the targeted release of glucocorticoids to immune cells [54,101]. By exploiting the biorthogonal property of the linkers, a phosphate diester was introduced into an alkyl chain. The drug conjugates underwent dual-enzymatic cleavage of pyrophosphatase and acid pyrophosphate to release the glucocorticoid (Figure 15). Nevertheless, as a proof of concept, it is significant for this linker to have been proven beyond merely in vitro laboratory analysis, using α-hCD70 and glucocorticoids as the delivery vehicle and payloads, respectively.
3.2. Non-Cleavable Linkers
Non-cleavable linkers are divided into two groups, namely thioether or maleimidocaproyl (MC). They consist of stable bonds that prevent proteolytic cleavage and ensure greater plasma stability than their cleavable counterparts. ADCs containing this type of linker depend on the complete lysosomal enzymatic degradation of the antibody to release payloads after internalization, resulting in the simultaneous detachment of the linker [93,102]. This linker strategy has been successfully explored by Genentech/Immunogen, with clinical approval of Trastuzumab emtansine (Kadcyla/T-DM1). This ADC contains a non-cleavable SMCC (N-succinimidyl-4-(maleimidomethyl) cyclohexane-1-carboxylate) linker connecting a warhead DM1 cytotoxin to Lys residues of anti-HER2 mAb Trastuzumab (Figure 16). This drug conjugate displayed greater activity than the conventional Trastuzumab-DM1, or Trastuzumab conjugated to other maytansinoids via reducible disulfide linkers [56]. Similarly, Monomethyl auristatin F (MMAF) drug conjugates with non-reducible thioether linkers were found to be more stable than Val–Cit conjugates and they also preserved their potency [51]. Of note, non-cleavable linkers allow for the alteration of the chemical properties of the small molecule in order to tune affinity for the transporter or improve potency [51,103]. The comparative advantage of non-cleavable linkers over their cleavable counterparts is their increased plasma stability [104]. Overall, non-cleavable linkers offer a greater therapeutic window than cleavable linkers since the payload derivative from non-cleavable ADCs can kill the target cells [51,103].
3.3. Conjugation Strategies
The success of an ADC depends mainly on the conjugation strategy. Two main types of conjugation are used in ADC design, namely chemical and enzymatic approaches. Most ADCs exploit the presence of Lys and Cys residues at the junction sites of the antibody, which can be modified for directional coupling. A typical IgG1 antibody molecule has roughly 90 Lys residues, of which approximately 30 can be modified for conjugation, implying that between 1 and 30 payloads can be covalently coupled to the antibody. The amine group of Lys and sulfhydryl of Cys are used for the chemical conjugation of the antibody-linker [77].
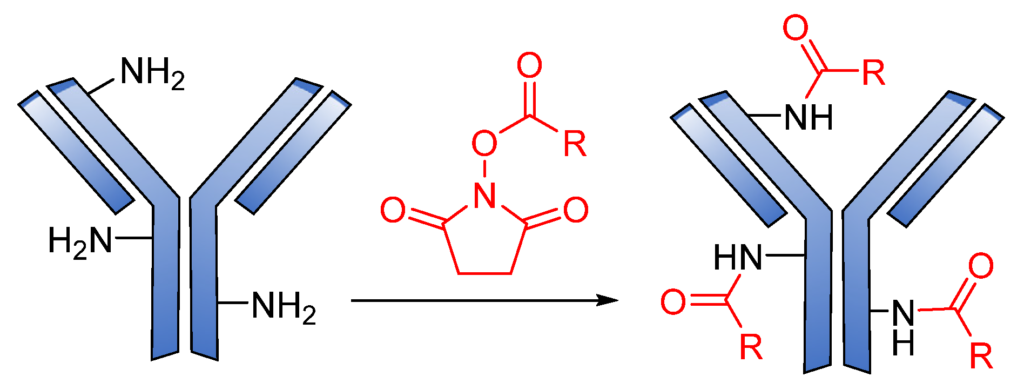
Generally, amide coupling is the method of choice for the chemical conjugation of payload and antibody Lys residues using activated carboxylic acid esters as linkers. This type of coupling gives a high-yielding ADC. The primary amine in Lys easily reacts with N-hydroxysuccinimide (NHS) esters introduced into the drug-linker, forming a stable amide, and a great number of commercial linkers rely on this method (Figure 17) [16,105].
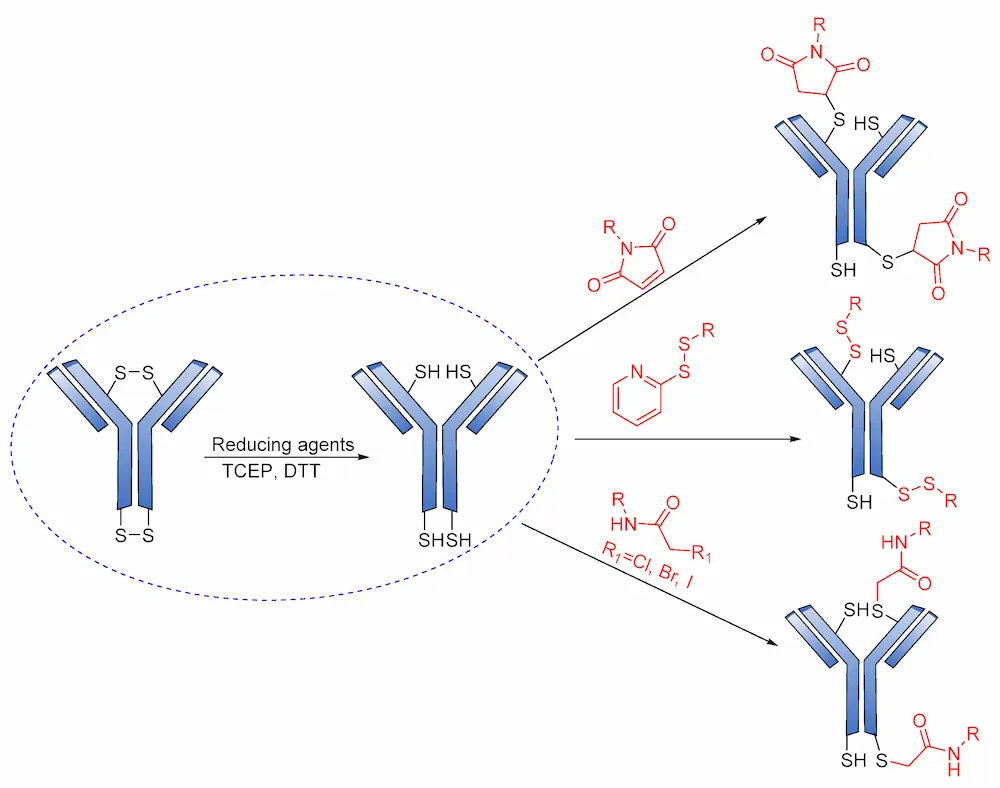
Cys are present in the antibodies and form disulfide bridges. Under careful reduction conditions, the disulfide bridge can be reduced by tris(2-carboxyethyl) phosphine (TCEP) or DL-dithiothreitol (DTT) to afford reactive thiol groups; meanwhile, intrachain disulfide bonds retain their unique state. The free thiol groups as attachment sites on the antibodies are then conjugated with a small linker molecule through various chemical reactions, such as Michael additions, disulfide formation, and a-halo carbonyl alkylations (Figure 18) [105].
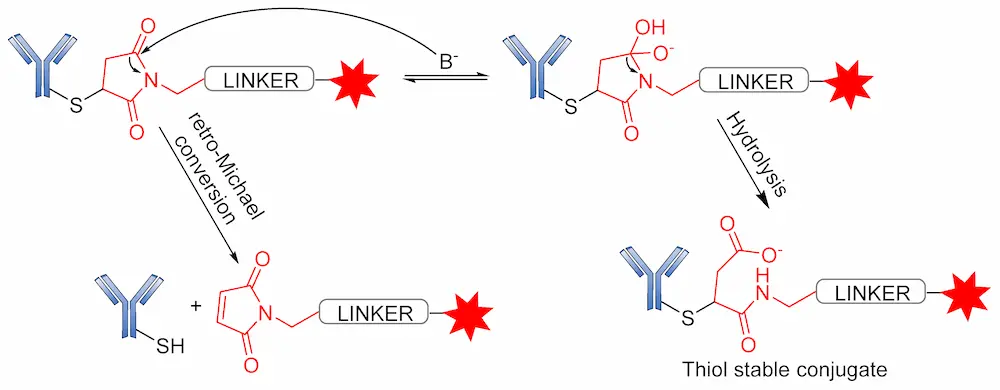
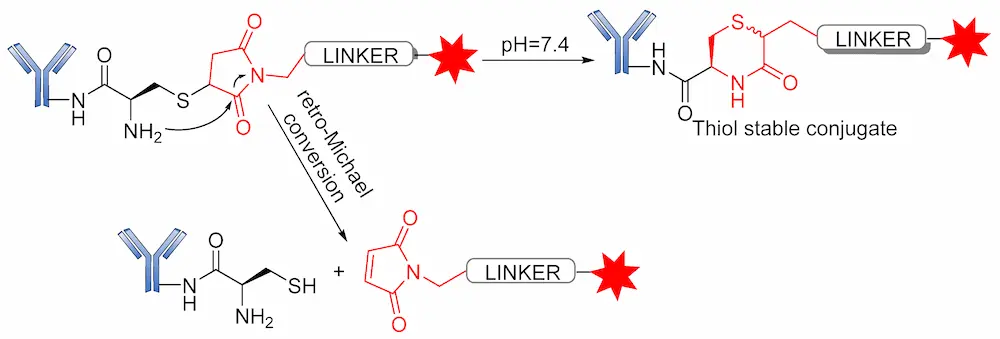
Several reports have revealed that a major challenge with the use of the maleimide-based strategy is its susceptibility to the premature release of payload through the retro–Michael reaction in the presence of blood thiols [105]. However, another reaction associated with the succinimide–thioether rings is hydrolysis, which converts the succinimide–thioether into succinic acid. This second side-reaction could help to prevent the retro-Michael reaction by firmly binding the conjugate to be thiol-stable (Figure 19) [106,107,108]. A new strategy recently developed by Lahnsteiner and coworkers to overcome the retro-Michael exchange is the formation of a stable 6-membered ring via transcyclization of the succinimide–thioether ring (Figure 20) [109].
4. Approved ADCs and ADCs in Clinical Trials
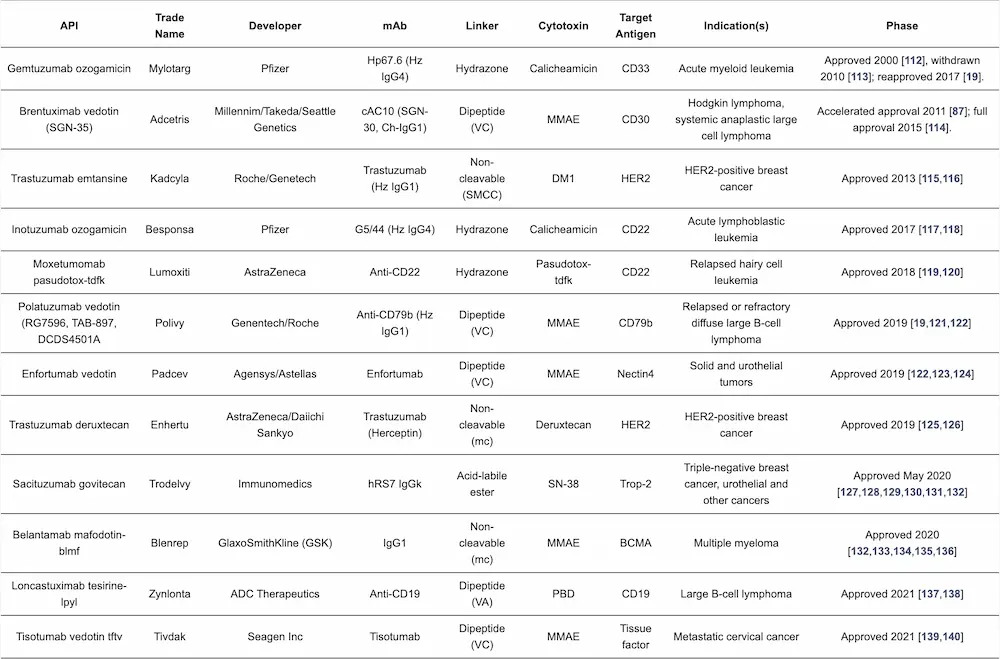
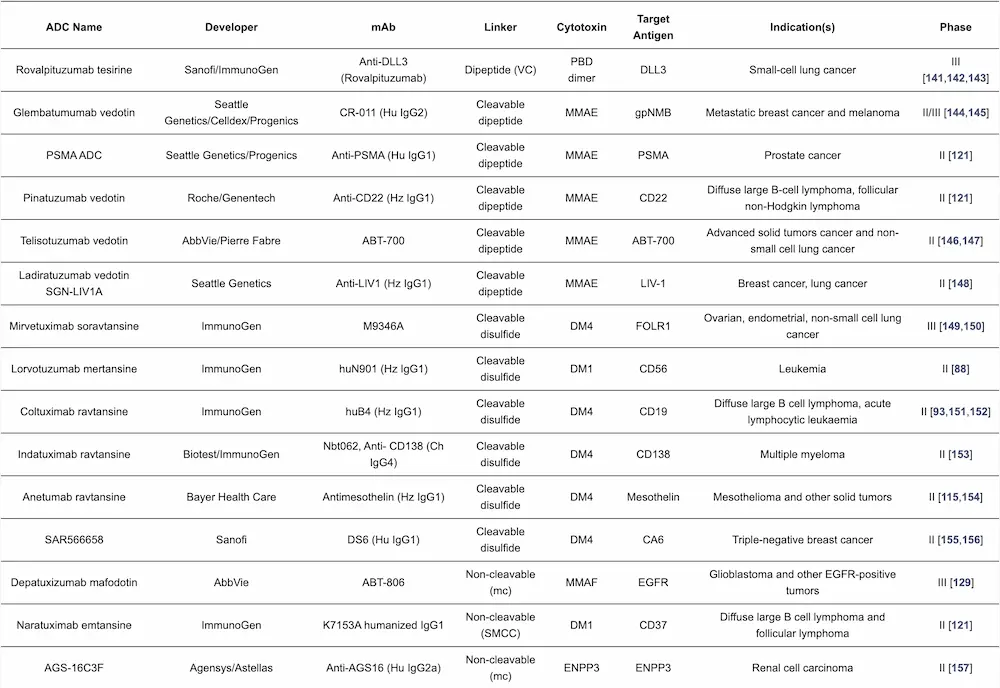
The importance of ADCs for cancer treatment is exemplified by the increasing number of these drugs on the market and in clinical studies for the treatment of solid tumors and hematologic malignancies [33,110]. There are currently 12 approved ADCs on the market (Table 2); however, considerable research efforts into the development of new ADCs are ongoing. Over 80 ADCs are now in clinical trials, and several others have been terminated due to linker instability and/or toxicity issues, among other factors [40,111]. The elements of approved ADCs are detailed in Table 2, while Table 3 covers selected examples of the ADCs in late-stage clinical trials.
5. ADC Mechanism of Action
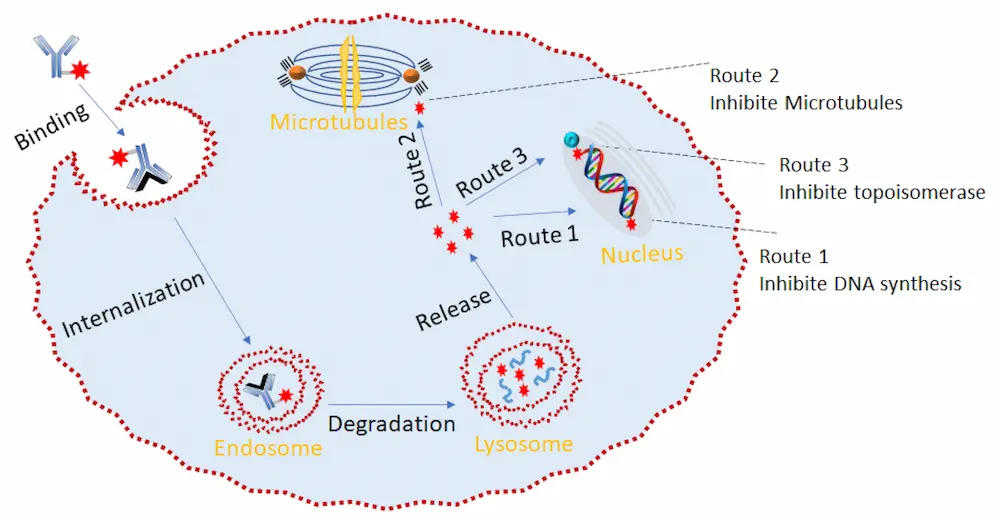
The mechanism of action of the ADC is shown in Figure 22. Relying on its ability to specifically recognize a well-expressed antigen, the ADC acts similarly to a shuttle that selectively delivers cytotoxic agents into the tumor cell via receptor-mediated endocytosis. Upon effective internalization of the ADC-antigen complex, it is fused with the endosome, which cleaves the complex, leading to the simultaneous recycling of the antigen and transport of the ADC to the lysosome. The ADC then undergoes lysosomal degradation to release the cytotoxin. This cytotoxin then binds to its target, leading to apoptosis or cell death via either DNA intercalation (route 1 and route 3) or binding to microtubulins (route 2) (Figure 13) [46]. Due to the complexity of the ADC internalization process, the localization of the tumor antigen on the cell surface is highly relevant to achieve efficient ADC binding [158].
6. Conclusions and Prospects
Ten ADCs have been approved by the FDA in the last five years (Gemtuzumab ozogamicin was first approved in 2000, withdrawn in 2010, and reapproved in 2017), which means that approximately 4% of all drugs approved during these years (240 drugs have been approved in total) are ADCs. However, the importance of ADCs in the context of the current toolbox to treat diseases is best exemplified by the large number that are in clinical phases. This pipeline assures that many more ADCs will be approved in the coming years, thereby fueling research in this field. In this regard, special attention must be given to the linker used. A suitable linker remains the mainstay of a successful ADC. In this context, a linker must remain stable in circulation and guarantee the safe release of the payload in the cell (such as release by restriction endocluease in lysosome or release after antibody degradation). Chemically cleavable (hydrazone, disulfide) linkers, enzymatically cleavable (peptide-based, β-glucuronide-based) linkers, and non-cleavable (thioether, maleimido caproyl) linkers are currently those most commonly used in ADC design. Given that linkers generally influence the stability, toxicity, pharmacokinetic properties, and pharmacodynamics of ADCs, considerable care must be taken in their selection for ADC design. In addition, the linker must take into consideration the reactive groups on the cytotoxic drugs, including the mAb and derivative functional groups.
Most ADCs in clinical stages use the common Lys and Cys residue motifs for their conjugation. In this context, researchers are currently directing significant effort toward studying new linkers. For instance, photo-sensitive ADC linkers and biorthogonal cleavable linkers are emerging classes that are still under study [126]. Although these linkers have some advantages, such as specificity, potency, and low toxicity, ADCs containing them are yet to gain regulatory approval.
Despite the huge progress made in the development ADCs, it is still difficult to postulate the exact market size of these pharmaceutical agents in the near future. However, it is envisaged that the development of new linkers will enlarge the design of new ADCs to further bolster the scope of oncology.
Final Word: Antibody Drug Conjugate Delivery
This article was lightly edited by Creative PEGWorks in adding an introductory paragraph.
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Author Contributions: Conceptualization, R.S., B.G.d.l.T. and F.A.; First Draft, R.S.; Final Review and Editing, R.S., B.G.d.l.T. and F.A. All authors have read and agreed to the published version of the manuscript.
Funding: The work was funded in part by the National Research Foundation (NRF) (#105892 and Blue Sky’s Research Program #120386).
Author Acknowledgements: The authors are grateful to the National Research Foundation (NRF) (#105892 and Blue Sky’s Research Program #120386). Rotimi Sheyi thanks MINTEK (South Africa) for a predoctoral fellowship to R.S.
References
- Ji, C.; Cheng, W.; Yuan, Q.; Müllen, K.; Yin, M. From Dyestuff chemistry to cancer theranostics: The rise of rylenecarboximides. Acc. Chem. Res. 2019, 52, 2266–2277. [Google Scholar] [CrossRef] [PubMed]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Zhou, Y. The role of surgery in the treatment of gastric cancer. J. Surg. Oncol. 2010, 101, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Hurwitz, M.D.; Krishnan, S.; Asea, A. Combined hyperthermia and radiotherapy for the treatment of cancer. Cancers 2011, 3, 3799–3823. [Google Scholar] [CrossRef]
- Garaci, E.; Pica, F.; Sinibaldi-Vallebona, P.; Pierimarchi, P.; Mastino, A.; Matteucci, C.; Rasi, G. Thymosin α1 in combination with cytokines and chemotherapy for the treatment of cancer. Int. Immu. Pharm. 2003, 3, 1145–1150. [Google Scholar] [CrossRef]
- Dan, N.; Setua, S.; Kashyap, V.K.; Khan, S.; Jaggi, M.; Yallapu, M.M.; Chauhan, S.C. Antibody-drug conjugates for cancer therapy: Chemistry to clinical implications. Pharmaceuticals 2018, 11, 32. [Google Scholar] [CrossRef] [Green Version]
- Rehman, K.; Tariq, M.; Akash, M.S.; Gillani, Z.; Qazi, M.H. Effect of HA 14-1 on Apoptosis-Regulating Proteins in HeLa Cells. Chem. Biol. Drug Des. 2014, 83, 317–323. [Google Scholar] [CrossRef]
- Chari, R.V.; Miller, M.L.; Widdison, W.C. Antibody–drug conjugates: An emerging concept in cancer therapy. Angew. Chem. 2014, 53, 3796–3827. [Google Scholar] [CrossRef]
- Kopp, A.; Thurber, G.M. Severing Ties: Quantifying the Payload Release from Antibody Drug Conjugates. Cell Chem. Biol. 2019, 26, 1631–1633. [Google Scholar] [CrossRef]
- Loadman, P. Anticancer drug development. Br. J. Cancer 2002, 86, 1665–1666. [Google Scholar] [CrossRef] [Green Version]
- DeVita, V.T.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mckertish, C.M.; Kayser, V. Advances and Limitations of Antibody Drug Conjugates for Cancer. Biomedicines 2021, 9, 872. [Google Scholar] [CrossRef] [PubMed]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ding, J.; Wang, Y.; Cheng, J.; Ji, S.; Zhuang, X.; Chen, X. Sequentially responsive shell-stacked nanoparticles for deep penetration into solid tumors. Adv. Mater. 2017, 29, 1701170. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Lorenzo, C.; Blanco-Fernandez, B.; Puga, A.M.; Concheiro, A. Crosslinked ionic polysaccharides for stimuli-sensitive drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 1148–1171. [Google Scholar] [CrossRef]
- Zhao, H.; Li, L.; Zheng, C.; Hao, Y.; Niu, M.; Hu, Y.; Chang, J.; Zhang, Z.; Wang, L. An intelligent dual stimuli-responsive photosensitizer delivery system with O2-supplying for efficient photodynamic therapy. Colloids Surf. B Biointerfaces 2018, 167, 299–309. [Google Scholar] [CrossRef]
- Xu, F.; Zhong, H.; Chang, Y.; Li, D.; Jin, H.; Zhang, M.; Wang, H.; Jiang, C.; Shen, Y.; Huang, Y. Targeting death receptors for drug-resistant cancer therapy: Codelivery of pTRAIL and monensin using dual-targeting and stimuli-responsive self-assembling nanocomposites. Biomaterials 2018, 158, 56–73. [Google Scholar] [CrossRef]
- Aghebati-Maleki, L.; Shabani, M.; Baradaran, B.; Motallebnezhad, M.; Majidi, J.; Yousefi, M. Receptor tyrosine kinase-like orphan receptor 1 (ROR-1): An emerging target for diagnosis and therapy of chronic lymphocytic leukemia. Biomed. Pharmacother. 2017, 88, 814–822. [Google Scholar] [CrossRef]
- Amani, N.; Dorkoosh, F.A.; Mobedi, H. ADCs, as novel revolutionary weapons for providing a step forward in targeted therapy of malignancies. Curr. Drug Deliv. 2020, 17, 23–51. [Google Scholar] [CrossRef]
- Liu, H.; Bolleddula, J.; Nichols, A.; Tang, L.; Zhao, Z.; Prakash, C. Metabolism of bioconjugate therapeutics: Why, when, and how? Drug Metab. Rev. 2020, 52, 66–124. [Google Scholar] [CrossRef]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M.; Dhimolea, E. The future of antibodies as cancer drugs. Drug Discov. Today 2012, 17, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Trail, P.A. Antibody drug conjugates as cancer therapeutics. Antibodies 2013, 2, 113–129. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.M. Drug-conjugated antibodies for the treatment of cancer. Br. J. Clin. Pharmacol. 2013, 76, 248–262. [Google Scholar] [CrossRef]
- Perez, H.L.; Cardarelli, P.M.; Deshpande, S.; Gangwar, S.; Schroeder, G.M.; Vite, G.D.; Borzilleri, R.M. Antibody–drug conjugates: Current status and future directions. Drug Discov. Today 2014, 19, 869–881. [Google Scholar] [CrossRef]
- Drake, P.M.; Rabuka, D. Recent developments in ADC technology: Preclinical studies signal future clinical trends. BioDrugs 2017, 31, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody–drug conjugates: The last decade. Pharm. J. 2020, 13, 245. [Google Scholar] [CrossRef]
- Lambert, J.M. Antibody–drug conjugates (ADCs): Magic bullets at last! Mol. Pharm. 2015, 12, 1701–1702. [Google Scholar] [CrossRef]
- Drachman, J.G.; Senter, P.D. Antibody-drug conjugates: The chemistry behind empowering antibodies to fight cancer. Hematol. Am. Soc. Hematol. Educ. Program Book 2013, 2013, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody–drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315. [Google Scholar] [CrossRef] [PubMed]
- Shor, B.; Gerber, H.-P.; Sapra, P. Preclinical and clinical development of inotuzumab-ozogamicin in hematological malignancies. Mol. Immunol. 2015, 67, 107–116. [Google Scholar] [CrossRef]
- Hughes, B. Antibody–drug conjugates for cancer: Poised to deliver? Nat. Rev. Drug Discov. 2010, 9, 665–667. [Google Scholar] [CrossRef]
- Carter, P.J.; Senter, P.D. Antibody-drug conjugates for cancer therapy. Cancer J. 2008, 14, 154–169. [Google Scholar] [CrossRef]
- Casi, G.; Neri, D. Antibody–drug conjugates: Basic concepts, examples and future perspectives. J. Control Release 2012, 161, 422–428. [Google Scholar] [CrossRef]
- Iyer, U.; Kadambi, V. Antibody drug conjugates—Trojan horses in the war on cancer. Toxicol. Appl. Pharmacol. 2011, 64, 207–212. [Google Scholar] [CrossRef]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates—An emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef]
- Donaghy, H. In Effects of Antibody, Drug and Linker on the Preclinical and Clinical Toxicities of Antibody-Drug Conjugates, mAbs; Taylor & Francis: Abingdon, UK, 2016; pp. 659–671. [Google Scholar]
- Lambert, J.M.; van Delft, F. Introduction to Antibody–Drug Conjugates. Drug Discov. 2021, 1–31. [Google Scholar] [CrossRef]
- Lee, P.; Kim, C.-U.; Seo, S.H.; Kim, D.-J. Current status of COVID-19 vaccine development: Focusing on antigen design and clinical trials on later stages. Immune Netw. 2021, 21, e4. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, G.; Tang, T.; Liang, T. Identification of tumor antigens and immune subtypes of pancreatic adenocarcinoma for mRNA vaccine development. Mol. Cancer 2021, 20, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Davey, A.S.; Call, M.E.; Call, M.J. The influence of chimeric antigen receptor structural domains on clinical outcomes and associated toxicities. Cancers 2021, 13, 38. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, H.; Viskov, C.; Garcia-Echeverria, C. Antibody–drug conjugates—A new wave of cancer drugs. Bioorg. Med. Chem. Lett. 2014, 24, 5357–5363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapra, P.; Shor, B. Monoclonal antibody-based therapies in cancer: Advances and challenges. Pharmacol. Ther. 2013, 138, 452–469. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Brown, S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef] [Green Version]
- Senter, P.D. Potent antibody drug conjugates for cancer therapy. Curr. Opin. Chem. Biol. 2009, 13, 235–244. [Google Scholar] [CrossRef]
- Flygare, J.A.; Pillow, T.H.; Aristoff, P. Antibody-drug conjugates for the treatment of cancer. Chem. Biol. Drug Des. 2013, 81, 113–121. [Google Scholar] [CrossRef]
- Chudasama, V.; Maruani, A.; Caddick, S. Recent advances in the construction of antibody–drug conjugates. Nat. Chem. 2016, 8, 114–119. [Google Scholar] [CrossRef]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: Effects of linker technology on efficacy and toxicity. Bioconjugate Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef]
- Nolting, B. Linker technologies for antibody–drug conjugates. In Antibody-Drug Conjugates; Springer: Berlin/Heidelberg, Germany, 2013; pp. 71–100. [Google Scholar]
- Zhao, R.Y.; Wilhelm, S.D.; Audette, C.; Jones, G.; Leece, B.A.; Lazar, A.C.; Goldmacher, V.S.; Singh, R.; Kovtun, Y.; Widdison, W.C. Synthesis and evaluation of hydrophilic linkers for antibody–maytansinoid conjugates. J. Med. Chem. 2011, 54, 3606–3623. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.C.; Cancilla, M.; Dooney, D.; Kwasnjuk, K.; Zhang, R.; Beaumont, M.; Figueroa, I.; Hsieh, S.; Liang, L.; Tomazela, D. Discovery of pyrophosphate diesters as tunable, soluble, and bioorthogonal linkers for site-specific antibody–drug conjugates. J. Am. Chem. Soc. 2016, 138, 1430–1445. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.D.L.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody–cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, D.; Grandjean, C.; Rousselot-Pailley, P.; Joly, P.; Bourel-Bonnet, L.; Santraine, V.; Gras-Masse, H.; Melnyk, O. Solid-phase functionalization of peptides by an α-hydrazinoacetyl group. J. Org. Chem. 2003, 68, 7033–7040. [Google Scholar] [CrossRef]
- Hamann, P.R.; Hinman, L.M.; Beyer, C.F.; Lindh, D.; Upeslacis, J.; Flowers, D.A.; Bernstein, I. An anti-CD33 antibody−calicheamicin conjugate for treatment of acute myeloid leukemia. Choice of linker. Bioconjugate Chem. 2002, 13, 40–46. [Google Scholar] [CrossRef]
- Kalia, J.; Raines, R.T. Hydrolytic stability of hydrazones and oximes. Angew. Chem. Int. Ed. 2008, 47, 7523–7526. [Google Scholar] [CrossRef] [Green Version]
- Melnyk, O.; Fehrentz, J.A.; Martinez, J.; Gras-Masse, H. Functionalization of peptides and proteins by aldehyde or keto groups. Pept. Sci. 2000, 55, 165–186. [Google Scholar] [CrossRef]
- Yang, J.; Chen, H.; Vlahov, I.R.; Cheng, J.-X.; Low, P.S. Evaluation of disulfide reduction during receptor-mediated endocytosis by using FRET imaging. Proc. Natl. Acad. Sci. USA 2006, 103, 13872–13877. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, S.; Luo, T.; Wang, C.; Zhao, J. Disulfide linkage: A potent strategy in tumor-targeting drug discovery. Curr. Med. Chem. 2012, 19, 2976–2983. [Google Scholar] [CrossRef]
- Chari, R.V. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. Acc. Chem. Res. 2008, 41, 98–107. [Google Scholar] [CrossRef] [PubMed]
- DiJoseph, J.F.; Dougher, M.M.; Evans, D.Y.; Zhou, B.-B.; Damle, N.K. Preclinical anti-tumor activity of antibody-targeted chemotherapy with CMC-544 (inotuzumab ozogamicin), a CD22-specific immunoconjugate of calicheamicin, compared with non-targeted combination chemotherapy with CVP or CHOP. Cancer Chemother. Pharmacol. 2011, 67, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Dubowchik, G.M.; Mosure, K.; Knipe, J.O.; Firestone, R.A. Cathepsin B-sensitive dipeptide prodrugs. 2. Models of anticancer drugs paclitaxel (Taxol®), mitomycin C and doxorubicin. Bioorg. Med. Chem. Lett. 1998, 8, 3347–3352. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Firestone, R.A. Cathepsin B-sensitive dipeptide prodrugs. 1. A model study of structural requirements for efficient release of doxorubicin. Bioorg. Med. Chem. Lett. 1998, 8, 3341–3346. [Google Scholar] [CrossRef]
- Koblinski, J.E.; Ahram, M.; Sloane, B.F. Unraveling the role of proteases in cancer. Clin. Chim. Acta 2000, 291, 113–135. [Google Scholar] [CrossRef]
- Albin, N.; Massaad, L.; Toussaint, C.; Mathieu, M.-C.; Morizet, J.; Parise, O.; Gouyette, A.; Chabot, G.G. Main drug-metabolizing enzyme systems in human breast tumors and peritumoral tissues. Cancer Res. 1993, 53, 3541–3546. [Google Scholar] [PubMed]
- Tranoy-Opalinski, I.; Legigan, T.; Barat, R.; Clarhaut, J.; Thomas, M.; Renoux, B.; Papot, S. β-Glucuronidase-responsive prodrugs for selective cancer chemotherapy: An update. Eur. J. Med. Chem. 2014, 74, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody–drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Fuselier, J.A.; Sun, L.; Woltering, S.N.; Murphy, W.A.; Vasilevich, N.; Coy, D.H. An adjustable release rate linking strategy for cytotoxin–peptide conjugates. Bioorg. Med. Chem. Lett. 2003, 13, 799–803. [Google Scholar] [CrossRef]
- Patterson, L.H.; McKeown, S.R.; Robson, T.; Gallagher, R.; Raleigh, S.M.; Orr, S. Antitumour prodrug development using cytochrome P450 (CYP) mediated activation. Anti-Cancer Drug Des. 1999, 14, 473–486. [Google Scholar]
- Coin, I.; Beyermann, M.; Bienert, M. Solid-phase peptide synthesis: From standard procedures to the synthesis of difficult sequences. Nat. Protoc. 2007, 2, 3247–3256. [Google Scholar] [CrossRef] [PubMed]
- Firestone, R.; Willner, D.; Hofstead, S.; King, H.; Kaneko, T.; Braslawsky, G.; Greenfield, R.; Trail, P.; Lasch, S.; Henderson, A. Synthesis and antitumor activity of the immunoconjugate BR96-Dox. J. Control Release 1996, 39, 251–259. [Google Scholar] [CrossRef]
- Saleh, M.; Abbott, S.; Perron, V.; Lauzon, C.; Penney, C.; Zacharie, B. Synthesis and antimicrobial activity of 2-fluorophenyl-4, 6-disubstituted [1,3,5] triazines. Bioorg. Med. Chem. Lett. 2010, 20, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Nie, Y.; Huse, W.D.; Watkins, J.D. Humanization of a murine monoclonal antibody by simultaneous optimization of framework and CDR residues. J. Mol. Biol. 1999, 294, 151–162. [Google Scholar] [CrossRef]
- Ducry, L.; Stump, B. Antibody−drug conjugates: Linking cytotoxic payloads to monoclonal antibodies. Bioconjugate Chem. 2010, 21, 5–13. [Google Scholar] [CrossRef]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Van der Velden, V.H.; te Marvelde, J.G.; Hoogeveen, P.G.; Bernstein, I.D.; Houtsmuller, A.B.; Berger, M.S.; van Dongen, J.J. Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: In vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood 2001, 97, 3197–3204. [Google Scholar] [CrossRef]
- Ravandi, F.; Estey, E.H.; Appelbaum, F.R.; Lo-Coco, F.; Schiffer, C.A.; Larson, R.A.; Burnett, A.K.; Kantarjian, H.M. Gemtuzumab ozogamicin: Time to resurrect? J. Clin. Oncol. 2012, 30, 3921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiJoseph, J.F.; Dougher, M.M.; Kalyandrug, L.B.; Armellino, D.C.; Boghaert, E.R.; Hamann, P.R.; Moran, J.K.; Damle, N.K. Antitumor efficacy of a combination of CMC-544 (inotuzumab ozogamicin), a CD22-targeted cytotoxic immunoconjugate of calicheamicin, and rituximab against non-Hodgkin’s B-cell lymphoma. Clin. Cancer Res. 2006, 12, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, A.; Shinjo, K.; Yamakage, N.; Ono, T.; Hirano, I.; Matsui, H.; Shigeno, K.; Nakamura, S.; Tobita, T.; Maekawa, M. CMC-544 (inotuzumab ozogamicin) shows less effect on multidrug resistant cells: Analyses in cell lines and cells from patients with B-cell chronic lymphocytic leukaemia and lymphoma. Br. J. Haematol. 2009, 146, 34–43. [Google Scholar] [CrossRef]
- Mthembu, S.N.; Sharma, A.; Albericio, F.; de la Torre, B.G. Breaking a Couple: Disulfide Reducing Agents. ChemBioChem 2020, 21, 1947–1954. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Fang, Y.-Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Wang, R.E.; Wang, F. Antibody-drug conjugates for non-oncological indications. Expert Opin. Biol. Ther. 2016, 16, 591–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCombs, J.R.; Owen, S.C. Antibody drug conjugates: Design and selection of linker, payload and conjugation chemistry. AAPS J. 2015, 17, 339–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellogg, B.A.; Garrett, L.; Kovtun, Y.; Lai, K.C.; Leece, B.; Miller, M.; Payne, G.; Steeves, R.; Whiteman, K.R.; Widdison, W. Disulfide-linked antibody−maytansinoid conjugates: Optimization of in vivo activity by varying the steric hindrance at carbon atoms adjacent to the disulfide linkage. Bioconjugate Chem. 2011, 22, 717–727. [Google Scholar] [CrossRef]
- Socinski, M.A.; Kaye, F.J.; Spigel, D.R.; Kudrik, F.J.; Ponce, S.; Ellis, P.M.; Majem, M.; Lorigan, P.; Gandhi, L.; Gutierrez, M.E. Phase 1/2 study of the CD56-targeting antibody-drug conjugate lorvotuzumab mertansine (IMGN901) in combination with carboplatin/etoposide in small-cell lung cancer patients with extensive-stage disease. Clin. Lung Cancer 2017, 18, 68–76.e2. [Google Scholar] [CrossRef] [PubMed]
- Gondi, C.S.; Rao, J.S. Cathepsin B as a cancer target. Expert Opin. Ther. Targets 2013, 17, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjugate Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef]
- Dorywalska, M.; Strop, P.; Melton-Witt, J.A.; Hasa-Moreno, A.; Farias, S.E.; Galindo Casas, M.; Delaria, K.; Lui, V.; Poulsen, K.; Loo, C. Effect of attachment site on stability of cleavable antibody drug conjugates. Bioconjugate Chem. 2015, 26, 650–659. [Google Scholar] [CrossRef]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase II study. J. Clin. Oncol. 2012, 30, 2190–2196. [Google Scholar] [CrossRef] [Green Version]
- Younes, A.; Bartlett, N.L.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.L.; Forero-Torres, A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821. [Google Scholar] [CrossRef] [Green Version]
- Moskowitz, C.H.; Nademanee, A.; Masszi, T.; Agura, E.; Holowiecki, J.; Abidi, M.H.; Chen, A.I.; Stiff, P.; Gianni, A.M.; Carella, A. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 385, 1853–1862. [Google Scholar] [CrossRef]
- Stephan, J.P.; Kozak, K.R.; Wong, W.L.T. Challenges in developing bioanalytical assays for characterization of antibody–drug conjugates. Bioanalysis 2011, 3, 677–700. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Isse, K.; Fujihira, T.; Takenoyama, M.; Saunders, L.; Bheddah, S.; Nakanishi, Y.; Okamoto, I. Prevalence of Delta-like protein 3 expression in patients with small cell lung cancer. Lung Cancer 2018, 115, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Caculitan, N.G.; Ma, Y.; Zhang, D.; Kozak, K.R.; Liu, Y.; Pillow, T.H.; Sadowsky, J.; Cheung, T.K.; Phung, Q.; Haley, B. Cathepsin B is dispensable for cellular processing of cathepsin B-cleavable antibody–drug conjugates. Cancer Res. 2017, 77, 7027–7037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffrey, S.C.; Andreyka, J.B.; Bernhardt, S.X.; Kissler, K.M.; Kline, T.; Lenox, J.S.; Moser, R.F.; Nguyen, M.T.; Okeley, N.M.; Stone, I.J. Development and properties of β-glucuronide linkers for monoclonal antibody−drug conjugates. Bioconjugate Chem. 2006, 17, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, S.C.; De Brabander, J.; Miyamoto, J.; Senter, P.D. Expanded utility of the β-glucuronide linker: ADCs that deliver phenolic cytotoxic agents. ACS Med. Chem. Lett. 2010, 1, 277–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolodych, S.; Michel, C.; Delacroix, S.; Koniev, O.; Ehkirch, A.; Eberova, J.; Cianférani, S.; Renoux, B.; Krezel, W.; Poinot, P. Development and evaluation of β-galactosidase-sensitive antibody-drug conjugates. Eur. J. Med. Chem. 2017, 142, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.C.; Dooney, D.; Zhang, R.; Liang, L.; Brandish, P.E.; Cheng, M.; Feng, G.; Beck, A.; Bresson, D.; Firdos, J. Novel phosphate modified cathepsin B linkers: Improving aqueous solubility and enhancing payload scope of ADCs. Bioconjugate Chem. 2016, 27, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Erickson, H.K.; Park, P.U.; Widdison, W.C.; Kovtun, Y.V.; Garrett, L.M.; Hoffman, K.; Lutz, R.J.; Goldmacher, V.S.; Blättler, W.A. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006, 66, 4426–4433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Paula Costa Monteiro, I.; Madureira, P.; De Vasconscelos, A.; Humberto Pozza, D.; Andrade de Mello, R. Targeting HER family in HER2-positive metastatic breast cancer: Potential biomarkers and novel targeted therapies. Pharmacogenomics 2015, 16, 257–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Linkers having a crucial role in antibody–drug conjugates. Int. J. Mol. Sci. 2016, 17, 561. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Chen, Y.; Wang, J.; Zheng, L.; Hutchinson, M.; Persson, J.; Ji, J. Characterization of ring-opening reaction of succinimide linkers in ADCs. J. Pharm. Sci. 2019, 108, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumey, L.N.; Charati, M.; He, T.; Sousa, E.; Ma, D.; Han, X.; Clark, T.; Casavant, J.; Loganzo, F.; Barletta, F. Mild method for succinimide hydrolysis on ADCs: Impact on ADC potency, stability, exposure, and efficacy. Bioconjugate Chem. 2014, 25, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.-Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.-F.; Mai, E. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012, 30, 184–189. [Google Scholar] [CrossRef]
- Lahnsteiner, M.; Kastner, A.; Mayr, J.; Roller, A.; Keppler, B.K.; Kowol, C.R. Improving the Stability of Maleimide–Thiol Conjugation for Drug Targeting. Chem. Eur. J. 2020, 26, 15867. [Google Scholar] [CrossRef]
- Birrer, M.J.; Moore, K.N.; Betella, I.; Bates, R.C. Antibody-drug conjugate-based therapeutics: State of the science. JNCI J. Natl. Cancer Inst. 2019, 111, 538–549. [Google Scholar] [CrossRef]
- Coats, S.; Williams, M.; Kebble, B.; Dixit, R.; Tseng, L.; Yao, N.-S.; Tice, D.A.; Soria, J.-C. Antibody–Drug Conjugates: Future Directions in Clinical and Translational Strategies to Improve the Therapeutic Index. Clin. Cancer Res. 2019, 25, 5441–5448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, E.L.; Larson, R.A.; Stadtmauer, E.A.; Estey, E.; Löwenberg, B.; Dombret, H.; Karanes, C.; Theobald, M.; Bennett, J.M.; Sherman, M.L. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J. Clin. Oncol. 2001, 19, 3244–3254. [Google Scholar] [CrossRef] [PubMed]
- Ricart, A.D. Antibody-drug conjugates of calicheamicin derivative: Gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin. Cancer Res. 2011, 17, 6417–6427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Trail, P.A.; Dubowchik, G.M.; Lowinger, T.B. Antibody drug conjugates for treatment of breast cancer: Novel targets and diverse approaches in ADC design. Pharmacol. Ther. 2018, 181, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Poon, K.A.; Flagella, K.; Beyer, J.; Tibbitts, J.; Kaur, S.; Saad, O.; Yi, J.-H.; Girish, S.; Dybdal, N.; Reynolds, T. Preclinical safety profile of trastuzumab emtansine (T-DM1): Mechanism of action of its cytotoxic component retained with improved tolerability. Toxicol. Appl. Pharmacol. 2013, 273, 298–313. [Google Scholar] [CrossRef] [Green Version]
- Sangha, R.; Davies, A.; Dang, N.H.; Ogura, M.; MacDonald, D.A.; Ananthakrishnan, R.; Paccagnella, M.L.; Vandendries, E.; Boni, J.; Goh, Y.T. Phase 1 study of inotuzumab ozogamicin combined with R-GDP for the treatment of patients with relapsed/refractory CD22+ B-cell non-Hodgkin lymphoma. J. Drug Assess. 2017, 6, 10–17. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, G.B.; Albericio, F. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2018, 23, 533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, S. Moxetumomab pasudotox: First global approval. Drugs 2018, 78, 1763–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreitman, R.J.; Dearden, C.E.; Zinzani, P.L.L.; Delgado, J.; Robak, T.; le Coutre, P.D.; Gjertsen, B.T.; Troussard, X.; Roboz, G.J.; Karlin, L. Moxetumomab Pasudotox-Tdfk in heavily pretreated patients with relapsed/refractory hairy cell leukemia (HCL): Long-term follow-up from the pivotal Phase 3 Trial. Blood 2019, 134, 2808. [Google Scholar] [CrossRef]
- Nejadmoghaddam, M.-R.; Minai-Tehrani, A.; Ghahremanzadeh, R.; Mahmoudi, M.; Dinarvand, R.; Zarnani, A.-H. Antibody-drug conjugates: Possibilities and challenges. Avi. J. Med. Bio. 2019, 11, 3. [Google Scholar]
- De la Torre, B.G.; Albericio, F. The pharmaceutical industry in 2019. An analysis of FDA drug approvals from the perspective of molecules. Molecules 2020, 25, 745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, E.; Weinstock, C.; Zhang, L.; Charlab, R.; Dorff, S.E.; Gong, Y.; Hsu, V.; Li, F.; Ricks, T.K.; Song, P. FDA approval summary: Enfortumab vedotin for locally advanced or metastatic urothelial carcinoma. Clin. Cancer Res. 2021, 27, 922–927. [Google Scholar] [CrossRef]
- Jain, R.K.; Skelton IV, W.P.; Zhang, J. Emerging treatment options for the treatment of metastatic urothelial cancer: Therapeutic potential of enfortumab vedotin. Cancer Manag. Res. 2020, 12, 8379. [Google Scholar] [CrossRef]
- Keam, S.J. Trastuzumab deruxtecan: First approval. Drugs 2020, 80, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Xiao, D.; Xie, F.; Liu, L.; Wang, Y.; Fan, S.; Zhou, X.; Li, S. Antibody—Drug conjugates: Recent advances in linker chemistry. Acta Pharm. Sin. B 2021, 11, 3889–3907. [Google Scholar] [CrossRef]
- Shvartsur, A.; Bonavida, B. Trop2 and its overexpression in cancers: Regulation and clinical/therapeutic implications. Genes Cancer 2015, 6, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starodub, A.N.; Ocean, A.J.; Shah, M.A.; Guarino, M.J.; Picozzi, V.J.; Vahdat, L.T.; Thomas, S.S.; Govindan, S.V.; Maliakal, P.P.; Wegener, W.A. First-in-human trial of a novel anti-Trop-2 antibody-SN-38 conjugate, sacituzumab govitecan, for the treatment of diverse metastatic solid tumors. Clin. Cancer Res. 2015, 21, 3870–3878. [Google Scholar] [CrossRef] [Green Version]
- Zeng, P.; Chen, M.-B.; Zhou, L.-N.; Tang, M.; Liu, C.-Y.; Lu, P.-H. Impact of TROP2 expression on prognosis in solid tumors: A Systematic Review and Meta-analysis. Sci. Rep. 2016, 6, 33658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardia, A.; Mayer, I.A.; Diamond, J.R.; Moroose, R.L.; Isakoff, S.J.; Starodub, A.N.; Shah, N.C.; O’Shaughnessy, J.; Kalinsky, K.; Guarino, M. Efficacy and safety of anti-trop-2 antibody drug conjugate sacituzumab govitecan (IMMU-132) in heavily pretreated patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2017, 35, 2141. [Google Scholar] [CrossRef] [PubMed]
- Heist, R.S.; Guarino, M.J.; Masters, G.; Purcell, W.T.; Starodub, A.N.; Horn, L.; Scheff, R.J.; Bardia, A.; Messersmith, W.A.; Berlin, J. Therapy of advanced non-small-cell lung cancer with an SN-38-anti-Trop-2 drug conjugate, sacituzumab govitecan. J. Clin. Oncol. 2017, 35, 2790–2797. [Google Scholar] [CrossRef] [PubMed]
- Torre, B.G.; Albericio, F. The pharmaceutical industry in 2020. An analysis of FDA drug approvals from the perspective of molecules. Molecules 2021, 26, 627. [Google Scholar] [CrossRef]
- Markham, A. Belantamab mafodotin: First approval. Drugs 2020, 80, 1607–1613. [Google Scholar] [CrossRef] [PubMed]
- Minor, C. Belantamab Mafodotin-Blmf (Blenrep). Oncology Times 2021, 43, 15. [Google Scholar] [CrossRef]
- Snyder, J. Belantamab Mafodotin-blmf: Management and Prevention of Ocular Toxicities. HOPA 2021, 14. [Google Scholar]
- Al Musaimi, O.; Al Shaer, D.; Albericio, F.; de la Torre, B.G. 2020 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2021, 14, 145. [Google Scholar] [CrossRef] [PubMed]
- Peng, H. Perspectives on the development of antibody-drug conjugates targeting ROR1 for hematological and solid cancers. Antib. Ther. 2021, 4, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Lee, A. Loncastuximab Tesirine: First Approval. Drugs 2021, 81, 1229–1233. [Google Scholar] [CrossRef]
- Markham, A. Tisotumab Vedotin: First Approval. Drugs 2021, 81, 2141–2147. [Google Scholar] [CrossRef]
- Tong, J.T.; Harris, P.W.; Brimble, M.A.; Kavianinia, I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef]
- Rudin, C.M.; Pietanza, M.C.; Bauer, T.M.; Ready, N.; Morgensztern, D.; Glisson, B.S.; Byers, L.A.; Johnson, M.L.; Burris, H.A., III; Robert, F. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: A first-in-human, first-in-class, open-label, phase 1 study. Lancet 2017, 18, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Moek, K.; de Groot, D.; de Vries, E.; Fehrmann, R. The antibody–drug conjugate target landscape across a broad range of tumour types. Ann. Oncol. 2017, 28, 3083–3091. [Google Scholar] [CrossRef]
- Pacheco, J.M.; Camidge, D.R. Antibody drug conjugates in thoracic malignancies. Lung Cancer 2018, 124, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Maric, G.; Annis, M.; Dong, Z.; Rose, A.; Ng, S.; Perkins, D.; MacDonald, P.; Ouellet, V.; Russo, C.; Siegel, P. GPNMB cooperates with neuropilin-1 to promote mammary tumor growth and engages integrin α 5 β 1 for efficient breast cancer metastasis. Oncogene 2015, 34, 5494–5504. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Barris, D.M.; Piperdi, S.; Kuo, V.; Everts, S.; Geller, D.; Houghton, P.; Kolb, E.A.; Hawthorne, T.; Gill, J. Targeting glycoprotein NMB with antibody-drug conjugate, glembatumumab vedotin, for the treatment of Osteosarcoma. Pediatr. Blood Cancer 2016, 63, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Angevin, E.; Spitaleri, G.; Rodon, J.; Dotti, K.; Isambert, N.; Salvagni, S.; Moreno, V.; Assadourian, S.; Gomez, C.; Harnois, M. A first-in-human phase I study of SAR125844, a selective MET tyrosine kinase inhibitor, in patients with advanced solid tumours with MET amplification. Eur. J. Cancer 2017, 87, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickler, J.H.; Weekes, C.D.; Nemunaitis, J.; Ramanathan, R.K.; Heist, R.S.; Morgensztern, D.; Angevin, E.; Bauer, T.M.; Yue, H.; Motwani, M. First-in-Human Phase I, Dose-Escalation and-Expansion Study of Telisotuzumab Vedotin, an Antibody-Drug Conjugate Targeting c-Met, in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2018, 78, 3298–3306. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Morgan, H.E.; Smart, K.; Zahari, N.M.; Pumford, S.; Ellis, I.O.; Robertson, J.F.; Nicholson, R.I. The emerging role of the LIV-1 Subfamily of Zinc Transporters in Breast Cancer. Mol. Med. 2007, 13, 396–406. [Google Scholar] [CrossRef]
- Moore, K.N.; Borghaei, H.; O’Malley, D.M.; Jeong, W.; Seward, S.M.; Bauer, T.M.; Perez, R.P.; Matulonis, U.A.; Running, K.L.; Zhang, X. Phase 1 dose-escalation study of mirvetuximab soravtansine (IMGN853), a folate receptor α-targeting antibody-drug conjugate, in patients with solid tumors. Cancer 2017, 123, 3080–3087. [Google Scholar] [CrossRef]
- Moore, K.N.; Martin, L.P.; O’Malley, D.M.; Matulonis, U.A.; Konner, J.A.; Perez, R.P.; Bauer, T.M.; Ruiz-Soto, R.; Birrer, M.J. Safety and activity of mirvetuximab soravtansine (IMGN853), a folate receptor alpha–targeting antibody–drug conjugate, in platinum-resistant ovarian, fallopian tube, or primary peritoneal cancer: A phase i expansion study. J. Clin. Oncol. 2017, 35, 1112. [Google Scholar] [CrossRef]
- Carol, H.; Szymanska, B.; Evans, K.; Boehm, I.; Houghton, P.J.; Smith, M.A.; Lock, R.B. The anti-CD19 antibody–drug conjugate SAR3419 prevents hematolymphoid relapse postinduction therapy in preclinical models of pediatric acute lymphoblastic leukemia. Clin. Cancer Res. 2013, 19, 1795–1805. [Google Scholar] [CrossRef] [Green Version]
- Ribrag, V.; Dupuis, J.; Tilly, H.; Morschhauser, F.; Laine, F.; Houot, R.; Haioun, C.; Copie, C.; Varga, A.; Lambert, J. A dose-escalation study of SAR3419, an anti-CD19 antibody maytansinoid conjugate, administered by intravenous infusion once weekly in patients with relapsed/refractory B-cell non-Hodgkin lymphoma. Clin. Cancer Res. 2014, 20, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Kelly, K.R.; Siegel, D.S.; Chanan-Khan, A.A.; Somlo, G.; Heffner, L.T.; Jagannath, S.; Zimmerman, T.; Munshi, N.C.; Madan, S.; Mohrbacher, A. Indatuximab ravtansine (BT062) in combination with low-dose dexamethasone and lenalidomide or pomalidomide: Clinical activity in patients with relapsed/refractory multiple myeloma. Blood 2016, 128, 4486. [Google Scholar] [CrossRef]
- Blumenschein, G.R.; Hassan, R.; Moore, K.N.; Santin, A.; Kindler, H.L.; Nemunaitis, J.J.; Seward, S.M.; Rajagopalan, P.; Walter, A.; Sarapa, N. Phase I study of anti-mesothelin antibody drug conjugate anetumab ravtansine (AR). J. Clin. Oncol. 2016, 34, 2509. [Google Scholar] [CrossRef]
- Boni, V.; Rixe, O.; Rasco, D.; Gomez-Roca, C.; Calvo, E.; Morris, J.C.; Tolcher, A.W.; Assadourian, S.; Guillemin, H.; Delord, J.-P. Abstract A73: A Phase I first-in-human (FIH) study of SAR566658, an anti CA6-antibody drug conjugate (ADC), in patients (Pts) with CA6-positive advanced solid tumors (STs)(NCT01156870). Clin. Trials 2013, 12, A73. [Google Scholar]
- Gomez-Roca, C.A.; Boni, V.; Moreno, V.; Morris, J.C.; Delord, J.-P.; Calvo, E.; Papadopoulos, K.P.; Rixe, O.; Cohen, P.; Tellier, A. A phase I study of SAR566658, an anti CA6-antibody drug conjugate (ADC), in patients (Pts) with CA6-positive advanced solid tumors (STs)(NCT01156870). J. Clin. Oncol. 2016, 34, 2511. [Google Scholar] [CrossRef]
- Thompson, J.A.; Motzer, R.J.; Molina, A.M.; Choueiri, T.K.; Heath, E.I.; Redman, B.G.; Sangha, R.S.; Ernst, D.S.; Pili, R.; Kim, S.K. Phase I Trials of Anti-ENPP3 Antibody–Drug Conjugates in Advanced Refractory Renal Cell Carcinomas. Clin. Cancer Res. 2018, 24, 4399–4406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parslow, A.C.; Parakh, S.; Lee, F.-T.; Gan, H.K.; Scott, A.M. Antibody–drug conjugates for cancer therapy. Biomedicines 2016, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Sheyi, R.; de la Torre, B.G.; Albericio, F. Linkers: An Assurance for Controlled Delivery of Antibody-Drug Conjugate. Pharmaceutics 2022, 14, 396. https://doi.org/10.3390/pharmaceutics14020396 [MDPI]